










| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website http://www.wjon.org |
Case Report
Volume 8, Number 3, June 2017, pages 81-85
Rare Presentation of Adrenocortical Carcinoma in a 4-Month-Old Boy
Sonali Malhotraa, e, Apoorva R. Waikarb, Prabhsimranjot Singhc, Ludovico Guarinid, Elka Jacobson-Dickmana, Roja Motaghedia, Irina Kazachkovaa
aDepartment of Pediatric Endocrinology, Infant and Children’s Hospital of Brooklyn, Maimonides Medical Center, Brooklyn, NY, USA
bDepartment of Pediatrics, Brooklyn Hospital Center, Brooklyn, NY, USA
cDepartment of Hematology Oncology, Maimonides Cancer Center, Brooklyn, NY, USA
dDepartment of Pediatric Hematology Oncology, Maimonides Medical Center, Brooklyn, NY, USA
eCorresponding Author: Sonali Malhotra, Department of Pediatric Endocrinology, Infant and Children’s Hospital of Brooklyn, Maimonides Medical Center, 977 48th Street, Brooklyn, NY 11219, USA
Manuscript accepted for publication May 10, 2017
Short title: Adrenocortical Carcinoma
doi: https://doi.org/10.14740/wjon1036w
| Abstract | ▴Top |
Adrenocortical carcinoma (ACC) is a rare malignancy and even rarer in infancy. Most of these tumors in pediatric age group are hormonally active and predominantly present with virilization. Cortisol hypersecretion presenting as Cushing syndrome is extremely rare and seen in older age groups. We report a 4-month-old infant who presented with linear growth arrest and excessive weight gain in early infancy, consequently diagnosed with ACC. On long-term follow-up for 7 years, he remained metastasis free following surgical resection and was not treated with chemotherapy.
Keywords: Adrenocortical carcinoma; Cushing syndrome; Virilizing tumors
| Introduction | ▴Top |
Adrenocortical tumors comprise 0.2% of the pediatric cancers [1]. Overall, 25 new cases are expected to occur annually in pediatric population in the United States with an estimated annual incidence of 0.2 - 0.3 cases per 1 million individuals [2]. The typical age of presentation is mostly during the early childhood with a median age of 3 - 4 years, but also there is a second, smaller peak during adolescence [3] (Table 1 [4-14]). On rare occasions, adrenocortical tumors have been reported in infancy, but due to relatively infrequent occurrence in this age group, information on the clinical course, long-term outcomes and prognosis is sparse.
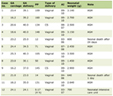 Click to view | Table 1. A Summary of All Case Reports of Pediatric ACC Along With Their Age at Diagnosis, Sex and Presenting Features That Were Found on PubMed |
| Case Report | ▴Top |
A 4-month-old male infant presented to the emergency department with a 3-day history of vomiting, diarrhea and difficulty breathing. Excessive and rapid weight gain was noted by the parents since the age of 2 months. The baby was previously healthy and born at term with a birth weight of 3.62 kg; the pregnancy and delivery were uncomplicated. There was no family history of consanguinity, malignancies, unexplained childhood deaths or stillbirth.
On a detailed physical exam, the baby was noted to have Cushingoid facies, poor muscle tone and excessive fat pad on the back and arms (Fig. 1). Notably, his length was 54 cm (below the 1st percentile) and the weight was disproportionately elevated at 6.7 kg (50th percentile). He was obese with weight for length ratio more than 99th percentile for the comparable age and sex. His blood pressure was elevated for age, ranging from 93 - 121 to 40 - 73 mm Hg. A large abdominal mass was palpable on the left lower quadrant and the borders were difficult to define. He had a prepubertal genital examination with no overt virilization; his phallus was typical in size but buried in the suprapubic fat pad. Neither acne, adult body odor, nor axillary hair was noted on examination.
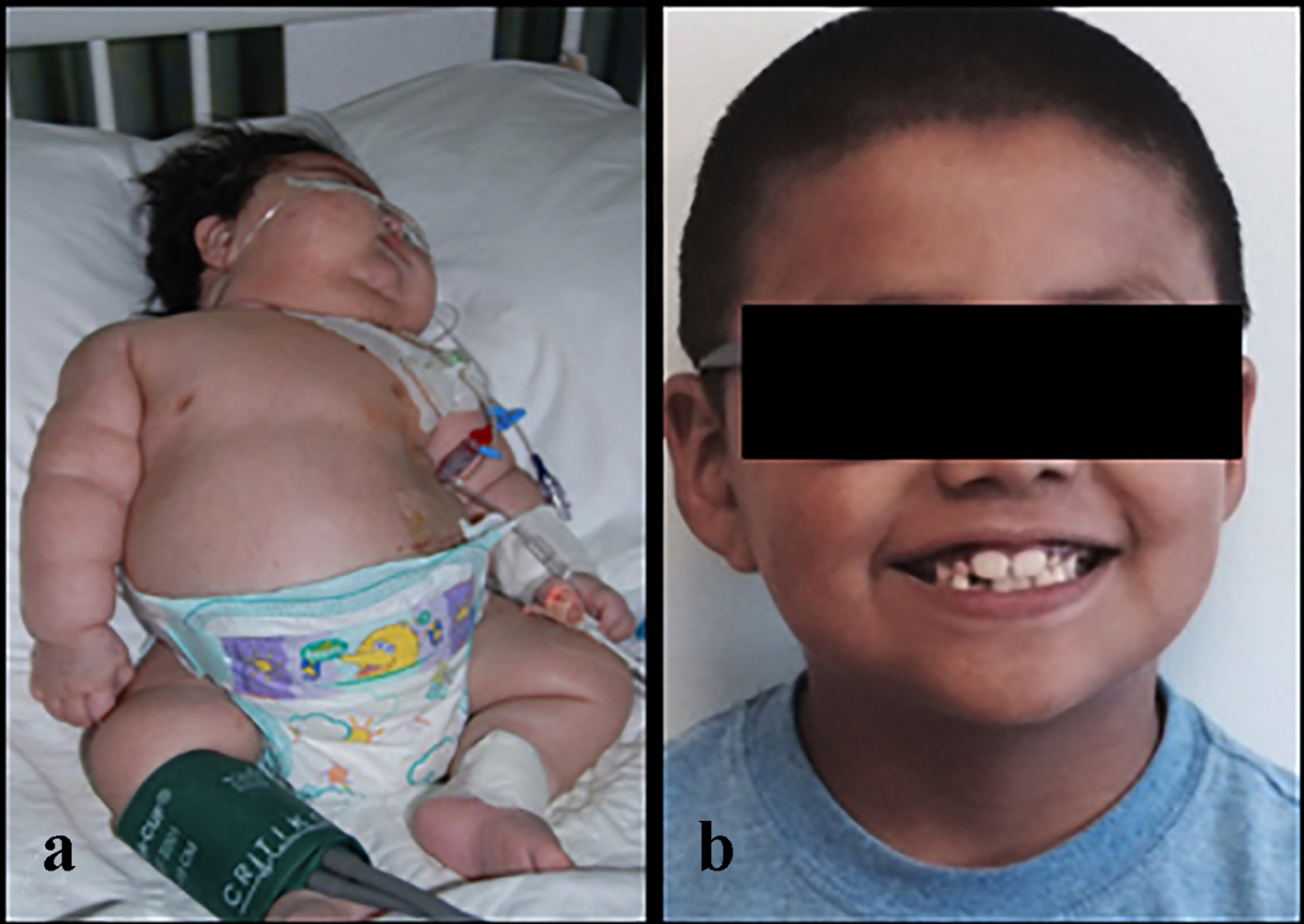 Click for large image | Figure 1. A 4-month-old male infant with an adrenocortical carcinoma at the time of presentation (a) and the same boy (b) at 7 years of age. |
In light of the respiratory distress, a chest radiograph was obtained and demonstrated bilateral upper lobe infiltrates, and henceforth he was admitted with a working diagnosis of pneumonia.
While being treated in the hospital for pneumonia, an evaluation was launched to delineate the etiology of the excessive weight gain, growth arrest, hypotonia and the abdominal mass. Laboratory evaluation revealed consistently elevated serum cortisol levels, irrespective of the time of the day with values of 73.0 μg/dL at 5:08 am and 68.6 μg/dL at 12:50 pm (normal range 4 - 22 μg/dL). Serum adrenocorticotropic hormone (ACTH) was suppressed at 7 pg/mL, at 5:16 am (normal range 0 - 60 pg/mL), suggestive of a primary adrenal cortisol hypersecretion. Dehydroepiandrosterone sulfate (DHEA-S) was mildly elevated at 115 μg/dL (normal range 16 - 96 μg/dL), but clinical features of hyperandrogenism were absent. The aldosterone level was normal at 23 ng/dL with a slightly elevated renin of 43.51 ng/mL/h. There was a slight increase in urine vanillylmandelic acid (VMA) and metanephrines, excluding a pheochromocytoma. An ultrasound demonstrated a 6.8 × 7.7 cm size solid mass at the upper pole of left kidney. A subsequent computerized tomography scan confirmed a large left-sided soft tissue mass with internal necrosis and a well-defined capsule abutting the left kidney with non-visualization of the left adrenal gland (Fig. 2). Henceforth, presumptive diagnosis of a functional cortisol producing adrenal tumor was made.
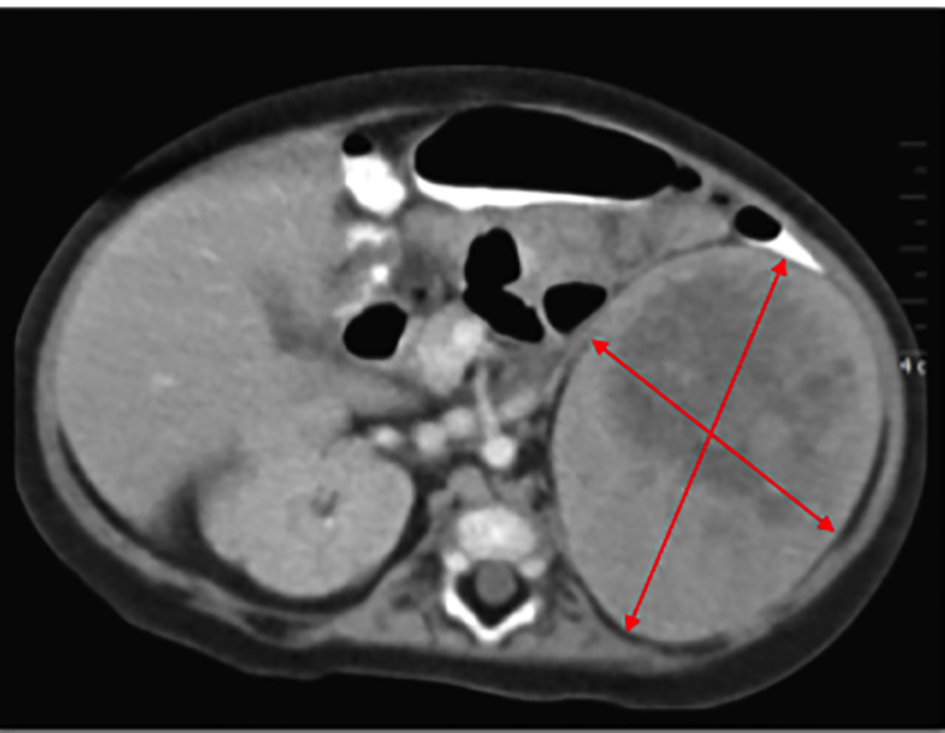 Click for large image | Figure 2. CT scan reports 6.8 × 7.7 cm size mass at the upper pole of left kidney. |
The baby underwent exploratory laparotomy and a left adrenalectomy was performed. Intraoperative findings revealed a 9.0 × 7.0 × 5.0 cm (315 cm3), well-encapsulated tumor weighing 180 g without breach of the capsule. The tumor was resected completely. There were no other signs of tumor spread including no liver metastasis or suspicious periaortic lymph nodes. Histological classification confirmed a high-grade adrenocortical carcinoma (24 mitotic figures/50 HPF) with a few pleomorphic nuclei (Fig. 3). The tumor was finally classified an adrenocortical carcinoma (ACC), stage 2 (based on combined American Joint Committee on Cancer (AJCC)/International Union Against Cancer (UICC) classification). During the perioperative period, the baby was treated with stress dosing of hydrocortisone and had an uneventful recovery. He continued to receive maintenance doses of hydrocortisone which were subsequently weaned. No adjuvant treatment with mitotane was done. During the recovery period, his linear growth velocity improved and his weight normalized (Fig. 4). Yearly follow-ups for the next 4 years and then most recently at 7 years and 10 months after treatment indicate continued health and successful recovery (Fig. 1).
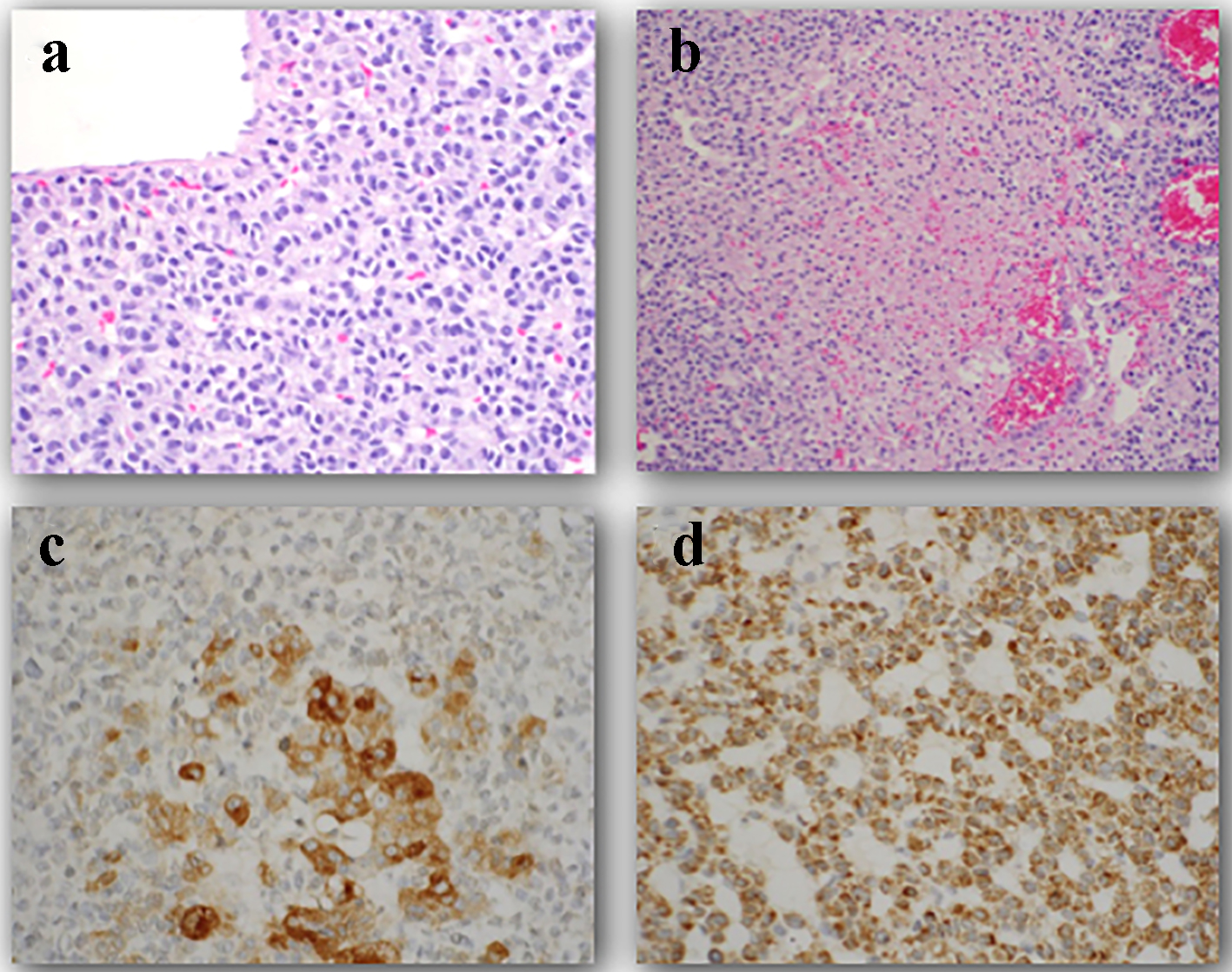 Click for large image | Figure 3. Histology of a high grade adrenocortical carcinoma in a 4-month-old male. (a) The tumor cells have large nuclei and prominent nucleoli. Furthermore, prominent mitotic figures are seen. (b) Confluent areas of necrosis are visualized. (c) Tumor cells show positive reaction for synaptophysin. (d) Tumor cells have positive reaction for melan-A. |
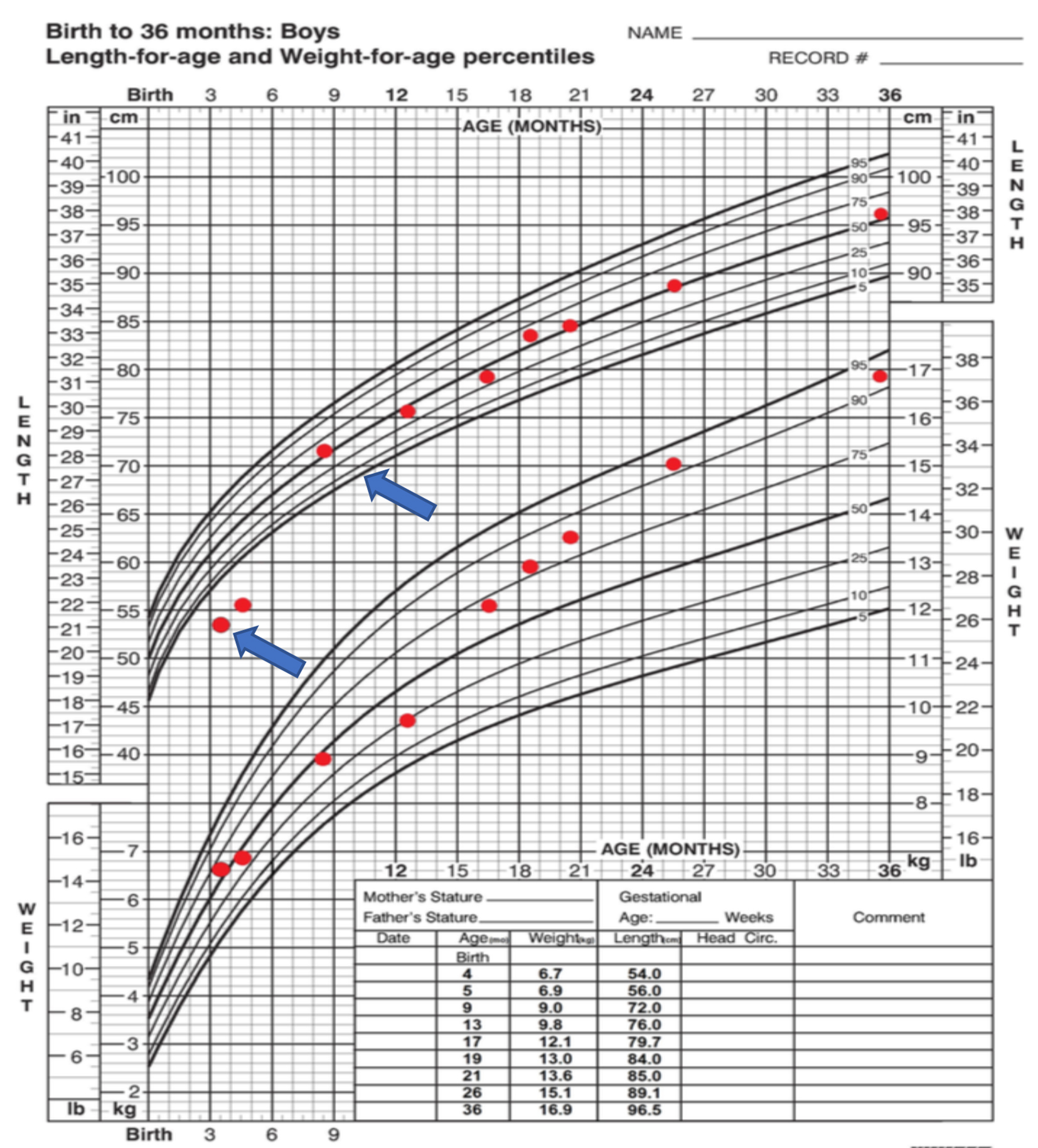 Click for large image | Figure 4. Growth chart demonstrating linear growth deceleration at diagnosis, followed by normal linear growth after surgical resection. |
| Discussion | ▴Top |
To date, there are only three well-described cases published in early infancy [4-6] but only one of which has information regarding long-term follow-up and prognosis. De Leon et al reported an affected male infant who was diagnosed at the age of 2 months with metastatic stage IV ACC who was treated with chemotherapy with mitotane for pulmonary metastasis and his 15-year course [6]. Herein, we report a second case of an infant with long-term follow-up who remained metastasis free following surgical resection and was not treated with chemotherapy.
Two national registries list infants with ACC but detailed case description is not offered [15, 16]. The Surveillance, Epidemiology and End Results (SEER) database for ACC in patients younger than 20 years of age, estimated an overall 5-year survival of 57% for all subjects. The SEER study also included subjects younger than 4 years, and estimated the 5-year survival for this younger group to be 91.1% [16]. The largest report by International Pediatric Adrenocortical Tumor Registry (IPACTR) included data from 254 pediatric patients with 18 infants younger than the age of 1 [15]. For all children, younger than 4 years with completely resected tumors weighing less than 200 g and without metastasis, the overall 5-year event free survival rate for infants was 91%. The report did not provide comments on outcomes of infantile ACC cases, specifically.
It is important to note that, unlike adults, in children the majority of adrenocortical cancers are functional with 80-90% having endocrine manifestations and, distinctively, virilization is reported to be present in > 80% of cases [2, 3]. However, in adults, adrenal hormone overproduction at presentation is reported to be around 40-60% and most commonly is hypercortisolism [17]. A presumptive explanation is that the fetal adrenal cortex has two different zones, with the outer definitive zone being steroidogenically latent until late gestation and the inner zone producing steroid hormones throughout gestation. The inner active fetal zone that is responsible for DHEAS makes up 85-90% of the total fetal adrenal cortex at birth and subsequently undergoes apoptosis thereafter [18]. Thus, it could explain the hormonally active nature of ACC in children, which, unlike our case, are more commonly androgen producing versus cortisol secreting. In the largest report by IPACTR of 254 children with adrenocortical tumors, isolated Cushing’s syndrome was found in only 5.5% of the patients with a median age of 12.6 years and henceforth tended to occur in the older children [15]. All 18 infants in this report had adrenal hormones overproduction [15]; however, the specific hormone functionality was not clearly elaborated in this report.
Isolated cortisol producing tumor or Cushing’s syndrome is extremely rare in general (5% of patients) [15]. After extensive medical literature review, our patient represents the fourth infantile ACC case, all of which presented with isolated Cushing’s syndrome. Whether isolated cortisol hypersecretion is more common in infantile ACC and even a proposed explanation for this phenomenon remains unclear.
Conclusion
ACC should be considered among patients with a history of rapid weight gain and growth failure, even in the absence of virilization. Based on the limited data on the long-term prognosis of ACC in infancy, this age range seems to have a favorable prognosis and in the few reported cases, early diagnosis and surgical excision appears to have provided cure. This appears consistent with International Pediatric Adrenocortical Tumor Registry (IPACTR) describing a favorable prognosis of ACC in younger age range with complete resection of tumors less than 200 g [15].
| References | ▴Top |
- Ribeiro RC, Figueiredo B. Childhood adrenocortical tumours. Eur J Cancer. 2004;40(8):1117-1126.
doi pubmed - Berstein L, Gurney JG. Carcinomas and other malignant epithelial neoplasms. In: Ries LA, Smith MA, Gurney JG, et al., eds. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub. No. 99-4649, Chapter 11. pp 139-148.
- Wieneke JA, Thompson LD, Heffess CS. Adrenal cortical neoplasms in the pediatric population: a clinicopathologic and immunophenotypic analysis of 83 patients. Am J Surg Pathol. 2003;27(7):867-881.
doi pubmed - Fudge EB, von Allmen D, Volmar KE, Calikoglu AS. Cushing syndrome in a 6-Month-Old infant due to Adrenocortical tumor. International Journal of Pediatric Endocrinology. 2009;2009:1-4.
doi pubmed - Garge S, Bawa M, Kanojia RP, Gupta K, Rao KL. A rare case of isolated Cushing syndrome in a 3-month-old boy. J Pediatr Endocrinol Metab. 2013;26(5-6):599-603.
doi pubmed - De Leon DD, Lange BJ, Walterhouse D, Moshang T. Long-term (15 years) outcome in an infant with metastatic adrenocortical carcinoma. J Clin Endocrinol Metab. 2002;87(10):4452-4456.
doi pubmed - Romaguera RL, Minagar A, Bruce JH, Jagid JR, Falcone S, Curless RG, Ragheb J, et al. Adrenocortical carcinoma with cerebral metastasis in a child: case report and review of the literature. Clin Neurol Neurosurg. 2001;103(1):46-50.
doi - Kanmaz T, Demirbilek S, Ozardali I, Safali M, Guran S, Yucesan S. Nonfunctioning adrenocortical carcinoma in a child. Pediatr Pathol Mol Med. 2003;22(5):405-410.
doi pubmed - Breidbart E, Cameo T, Garvin JH, Hibshoosh H, Oberfield SE. Pubertal outcome in a female with virilizing adrenocortical carcinoma. J Pediatr Endocrinol Metab. 2016;29(4):503-509.
doi pubmed - Kim MS, Yang EJ, Cho DH, Hwang PH, Lee DY. Virilizing adrenocortical carcinoma advancing to central precocious puberty after surgery. Korean J Fam Med. 2015;36(3):150-153.
doi pubmed - Arico M, Bossi G, Livieri C, Raiteri E, Severi F. Partial response after intensive chemotherapy for adrenal cortical carcinoma in a child. Med Pediatr Oncol. 1992;20(3):246-248.
doi pubmed - Wong LM, Li CH, Chan OK, Shek CC, Kwong NS. A 12-year-old chinese girl with Cushing syndrome and virilization due to adrenocortical carcinoma. J Pediatr Endocrinol Metab. 2011;24(3-4):193-196.
doi pubmed - Sorgo W, Meyer D, Rodens K, Homoki J, Heinze E, Heymer B, Siebenmann R. Testosterone-secreting adrenocortical tumor in a pubertal girl. Case report and review of the literature. Horm Res. 1988;30(6):217-223.
doi pubmed - Ghazizadeh F, Ebadi M, Alavi S, Arzanian M, Shamsian B, Jadali F. Adrenocortical carcinoma presenting with heterosexual pseudoprecocious puberty shortly after birth: case report and review. Ecancermedicalscience. 2013;7:289.
pubmed - Michalkiewicz E, Sandrini R, Figueiredo B, Miranda EC, Caran E, Oliveira-Filho AG, Marques R, et al. Clinical and outcome characteristics of children with adrenocortical tumors: a report from the International Pediatric Adrenocortical Tumor Registry. J Clin Oncol. 2004;22(5):838-845.
doi pubmed - McAteer JP, Huaco JA, Gow KW. Predictors of survival in pediatric adrenocortical carcinoma: a Surveillance, Epidemiology, and End Results (SEER) program study. J Pediatr Surg. 2013;48(5):1025-1031.
doi pubmed - Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, Jolly S, et al. Adrenocortical carcinoma. Endocr Rev. 2014;35(2):282-326.
doi pubmed - Coulter CL. Fetal adrenal development: insight gained from adrenal tumors. Trends Endocrinol Metab. 2005;16(5):235-242.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.