

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 13, Number 5, October 2022, pages 289-298

A ROS/Akt/NF-κB Signaling Cascade Mediates Epidermal Growth Factor-Induced Epithelial-Mesenchymal Transition and Invasion in Human Breast Cancer Cells
Wei Li Mina, f, g, Bao Feng Wangb, f, Bao Bao Lianga, Lun Zhangc, Ji Yuan Panb, Yi Huangd, Yang Zhaoa, Shuai Lina, Yi Han Zhaoe, Shu Qun Zhanga, Qing Yong Mac
aDepartment of Oncology, The Second Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710004, China
bDepartment of Radiation Therapy, Second Affiliated Hospital, Xi’an Jiaotong University, Xi’an 710004, China
cDepartment of Hepatobiliary Surgery, First Affiliated Hospital, Xi’an Jiaotong University, Xi’an 710061, China
dDepartment of Ultrasound, Xi’an Chest Hospital, Xi’an 710061, China
eSpecial Stomatology Department, College of Stomatology, Xi’an Jiaotong University, Xi’an 710004, China
fThese authors contributed equally to this work.
gCorresponding Author: Department of Oncology, The Second Affiliated Hospital of Xi’an Jiaotong University, Xi’an 710004, China
Manuscript submitted July 27, 2022, accepted August 23, 2022, published online October 22, 2022
Short title: ROS/Akt/NF-κB Mediates EMT and Invasion
doi: https://doi.org/10.14740/wjon1518
| Abstract | ▴Top |
Background: As one of the most widely used anti-diabetic drugs for type II diabetes, metformin has been shown to exhibit anti-cancer activity in recent years. Epidermal growth factor (EGF) and its receptor, EGFR, play important roles in cancer metastasis in various tumors, including breast cancer. Epithelial-mesenchymal transition (EMT) is a critical process for cancer invasion and metastasis. In this study, we use EGF as a metastatic inducer to investigate the effect of metformin on cancer cell migration, invasion and EMT.
Methods: Human breast cancer MCF-7 cells were exposed to EGF with or without metformin or N-acetyl cysteine (NAC). The effects of metformin on breast cancer cell proliferation were analyzed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. The production of reactive oxygen species (ROS) was tested using 2,7-dichlorodihydrofluorecein diacetate (DCFH-DA). The migratory and invasive abilities of tumor cells were analyzed using wound healing assay and transwell invasion assay, respectively. The expressions of E-cadherin, N-cadherin and Snail were tested using real-time quantitative polymerase chain reaction (qRT-PCR) and western blotting at mRNA and protein levels. The activation of protein kinase B (Akt) and nuclear factor kappa B (NF-κB) were measured by western blotting.
Results: Our results showed that metformin inhibited breast cancer cell proliferation in a dose-dependent manner with or without EGF. EGF-induced alterations in cell morphology that are characteristic of EMT were reversed by metformin. Metformin also inhibited the EGF-modulated expression of E-cadherin, N-cadherin and Snail and further suppressed cell invasion and migration. In addition, metformin suppressed EGF-induced phosphorylation of Akt and NF-κB. ROS is involved in EGF-induced cancer invasion and activation of phosphatidylinositol 3-kinase (PI3K)/Akt/NF-κB pathway.
Conclusion: Taken together, these data indicate that metformin suppresses EGF-induced breast cancer cell migration, invasion and EMT through the inhibition of the PI3K/Akt/NF-κB pathway. These results provide a novel mechanism to explain the role of metformin as a potent anti-metastatic agent in breast cancer cells.
Keywords: Metformin; EGF; EMT; PI3K/Akt; Breast cancer
| Introduction | ▴Top |
Breast cancer is one of the most highly malignant diseases afflicting women worldwide, with an estimated 246,660 new cases and 40,450 deaths in 2016 in the USA [1]. It is expected that breast cancer alone accounts for 29% of all new cancer diagnoses in women [1]. Several treatment decisions may be offered to women who suffer from breast cancer, including surgery, radiotherapy, endocrine and chemotherapy. However, due to lymphatic and hematogenous metastasis occurring at the early stage, breast cancer is still the primary leading cause of cancer deaths in women worldwide [2]. Chemotherapy is a pivotal therapeutic method for breast cancer patients, whereas the development of multi-drug resistance usually leads to the failure of chemotherapy [3]. Understanding the molecular basis of tumor metastasis is highly needed for developing new strategies to prevent and treat breast cancer.
Epithelial-to-mesenchymal transition (EMT), which is characterized by the loss of epithelial characteristics and the acquisition of mesenchymal morphology, has been recognized both as a physiological mechanism for embryonic development and wound healing, and as a pathological mechanism in tumor progression [4, 5]. A typical symbol of EMT is loss of epithelial markers (E-cadherin) and gain mesenchymal markers (vimentin and N-cadherin) as well as the up-regulation of associated transcriptional regulators (Snail, Twist) [6]. It is believed that EMT is an important event in the initial step of the tumor metastasis allowing cells to acquire migratory and invasive properties [4]. Thus, EMT is considered as a crucial factor in cancer progression. Blocking this process is a promising therapeutic strategy to limit cancer diffusion.
Growth factors and cytokines can promote EMT triggering specific signaling pathways [7, 8]. Epidermal growth factor (EGF), a low molecular weight (Mr = 6,045) polypeptide, is a well-known growth factor that induces cancer cell migration and invasion [7]. Some studies have indicated a role of EGF alone, or in combination with other growth factors, in disrupting cell-cell junctions and promoting EMT in breast tumor [9, 10]. EGF stimulates signaling pathways through the EGF receptor (EGFR), which is frequently over-expressed in breast cancers with a higher incidence of distant metastases and decreased survival [11]. The two major intracellular pathways activated by EGF/EGFR system are the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinases (ERK) cascade and the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway - all known for being frequently dysregulated in breast cancer [12].
Metformin (1,1-dimethylbiguanide hydrochloride), the most prescribed drug for type II diabetes mellitus, has received more and more attention as a potentially useful therapeutic agent for treating cancer in recent years [13]. Preclinical studies have shown that metformin is able to inhibit cell proliferation, induce cell apoptosis, suppress cell migration and invasion, reverse multidrug resistance, as well as enhance radiosensitivity in breast cancer [3, 14, 15]. Metformin exerts its anti-cancer action via adenosine monophosphate-activated protein kinase (AMPK)-dependent and/or AMPK-independent mechanisms. AMPK is linked with the PI3K/Akt and MAPK/ERK cascades [16].
Since EGF is a known inducer of EMT in breast cancer cells [7], in the present study, we tested the hypothesis that metformin is able to inhibit EGF-induced invasion and migration as well as EMT of MCF-7 cells through the inhibition of the PI3K/Akt pathway. Results from this study suggest that metformin is able to inhibit the action of EGF via inhibiting ROS production. Metformin treatment may be a novel option for therapy of breast cancer via the inhibition of the PI3K/Akt/NF-κB pathway.
| Materials and Methods | ▴Top |
Cell culture
The human breast cancer cell line, MCF-7, was obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin (GIBCO, Grand Island, NY, USA) at 37 °C in a 5% CO2 humidified atmosphere.
Reagents and antibodies
RPMI-1640 medium and FBS were from Gibco (Grand Island, NY, USA). Metformin, N-acetyl cysteine (NAC) and EGF were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies against E-cadherin, N-cadherin and Snail were procured from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-NF-κB, anti-phospho-NF-κB p65 (Ser468), anti-Akt and anti-phospho-Akt (Ser473) antibodies were obtained from Cell Signaling Technology (Beverly, MA, USA). The PI3K inhibitor LY294002 was obtained from Sigma Chemical Co. AG 1478 (EGFR inhibitor) was purchased from Calbiochem (San Diego, CA, USA). The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and DMSO were purchased from Sigma Chemical Company (St. Louis, MO, USA). Other reagents were purchased from common commercial sources.
MTT assays
MCF-7 cells were plated in triplicate at 1 × 104 cells per well in 96-well culture plates in culture media. The cells were then treated with different concentrations of metformin with or without EGF. After incubation for 24, 48, and 72 h, MTT solution was added to each well, and then the cells were incubated at 37 °C with 5% CO2 for 4 h. DMSO was then added to each well. The absorbance at 490 nm was measured using a spectrophotometer (Bio-Rad, CA, USA).
Morphologic analysis
Serum-starved cells were treated with metformin (10 mmol/L) followed by EGF (50 ng/mL) treated for 24 h. The cells were analyzed using a phase contrast microscope. The experiment was repeated in triplicate.
ROS determination
The level of intracellular ROS was determined by measuring the oxidative conversion of cell permeable 2,7-dichlorodihydrofluorecein diacetate (DCFH-DA) using the ROS assay kit. In brief, after treatment with DCFH-DA, the cells were washed three times with phosphate-buffered saline (PBS) and fluorescence intensity measured using a fluorometer system (BD Biosciences) at a wavelength pair of 488/525 nm.
Wound healing assay
MCF-7 cells were plated in 24-well plates and grown to 90-100% confluence. After serum starvation overnight, a wound line was created by scratching the cells with a sterile pipette tip. Cellular debris was removed by washing twice with PBS and then a medium containing EGF (50 ng/mL) was added with or without metformin (10 mmol/L) for 24 h. Images were taken at the time of 0 and 24 h under an inverted microscope (× 10). The relative distance traveled by the leading edge were counted in five random fields from each treatment and assessed using Photoshop software.
Transwell matrigel invasion assay
The invasion of MCF-7 cells was performed in transwell chambers using 8 µm transwell inserts (Millipore, Billerica, MA, USA) coated with 25 µL matrigel (BD, Bedford, MA, USA). In brief, the cells were seeded into the upper inserts (5 × 104) in the medium containing 1% FBS. Medium containing 20% FBS was placed in the lower chambers. The cells were treated with EGF (50 ng/mL) and/or metformin (10 mmol/L) and allowed to migrate for 48 h. At the end of incubation period, the non-invading cells on the upper surface of the membrane were removed with a wet cotton swab. Cells crossing the matrigel to the lower surface of the membrane were fixed and stained with crystal violet. The invading cells on the lower surface of the membrane filter were counted under an inverted microscope (× 20). The experiment was repeated in triplicate.
Real-time quantitative polymerase chain reaction (qRT-PCR)
Total RNA was isolated using the Fastgen200 RNA isolation system (Fastgen, Shanghai, China) and was reverse-transcribed into cDNA using the Fermentas RevertAidTM Kit (MBI Fermentas, Canada). The PCR conditions for E-cadherin, N-cadherin and Snail were 95 °C for 2 min, followed by 40 cycles of 95 °C for 0.5 min, 50 °C for 0.5 min and 72 °C for 0.5 min. The following primers were used: E-cadherin-F: 5’-GAA CGC ATT GCC ACA TAC AC-3’, E-cadherin-R: 5’-GAA TTC GGG CTT GTT GTC AT-3’, N-cadherin-F: 5’-TGT TTG ACT ATG AAG GCA GTG G-3’, N-cadherin-R: 5’-TCA GTC ATC ACC TCC ACC AT-3’, Snail-F: 5’-GCG AGC TGC AGG ACT CTA AT-3’, Snail-R: 5’-GGA CAG AGT CCC AGA TGA GC-3’, β-actin-F: 5’-CTT AGT TGC GTT ACA CCC TTT CTT G-3’, β-actin-R: 5’-CTG TCA CCT TCA CCG TTC CAG TTT-3’. β-actin was used as an internal control. The relative gene expression was calculated using the previously described 2-ΔΔCt method.
Western blotting
Equal amounts of protein (20 mg) were separated by SDS-polyacrylamide gel and electrotransferred onto nitrocellulose membranes. After blocking with 5% non-fat dry milk in Tris-buffered saline (TBS) for 2 h and washed with TBST buffer, membranes were probed with primary antibodies at 4 °C overnight and then incubated with secondary antibody for 2 h at 37 °C. The protein bands were detected using the ECL detection system (Pierce).
Statistical analysis
Statistical analysis was carried out using SPSS 17.0 software. Analysis of variance (ANOVA) was chosen to analyze statistical difference. The results were presented as the means ± standard deviation (SD). P < 0.05 was considered statistically significant. All experiments were repeated independently at least three times.
| Results | ▴Top |
Metformin inhibits EGF-induced proliferation and ROS production of MCF-7 cells
We first explored the effect of metformin on the cell viability of MCF-7 using the MTT assay. As shown in Figure 1a, MCF-7 cells were treated with metformin at various concentrations ranging from 0 to 20 mmol/L for 24, 48, and 72 h. The results proved that the proliferation of MCF-7 cells was decreased in response to the treatment of metformin in both time- and dose-dependent manners. Compared with the untreated control, cells treated with metformin at a concentration between 0 and 10 mmol/L exhibited no cytotoxicity, because the 50% inhibitory concentration (IC50) was nearly 10 mmol/L. Therefore, a treatment concentration of 10 mmol/L of metformin was applied in all subsequent experiments. EGF has been proven to promote the proliferation of breast cancer cells [17]. Here we found that the proliferation of MCF-7 cells treated with EGF was increased compared with the control group and this increase could be reduced in the presence of metformin in a dose-dependent manner (Fig. 1b). ROS generation is related with tumor progression. In this study, we also examined the effect of metformin on EGF-induced ROS generation in MCF-7 cells. Our data showed that, the generation of ROS was increased dramatically upon treatment with EGF from 0 to 180 min (Fig. 1c). The role of EGF-induced ROS production was further investigated by treatment with metformin or a scavenger of ROS, NAC. Treatment of cells with metformin or NAC prior to EGF diminished the production of ROS induced by EGF treatment (Fig. 1d).
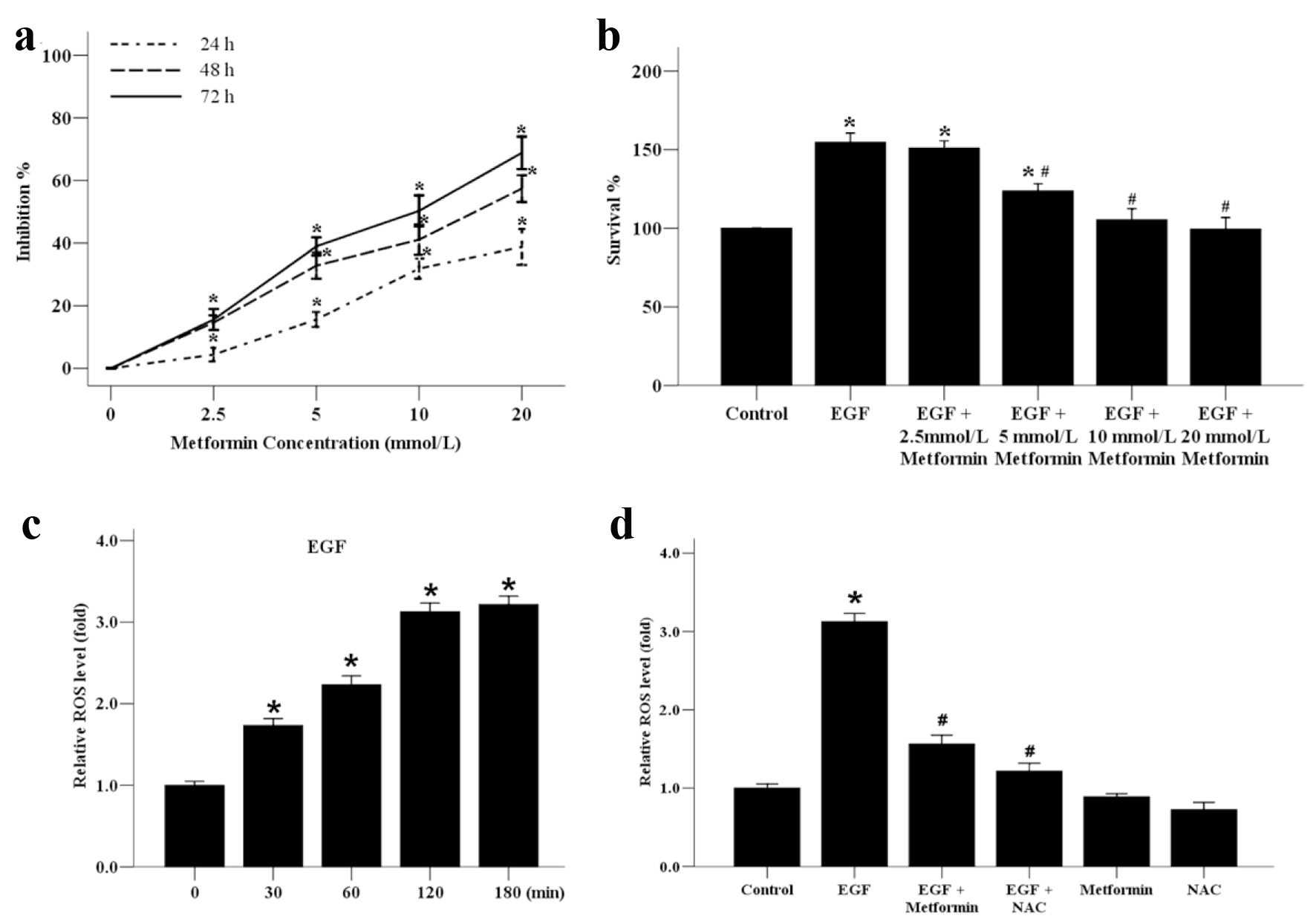 Click for large image | Figure 1. The effect of metformin on the growth and ROS generation of the human MCF-7 breast cancer cells. (a) MCF-7 cells were incubated with various concentrations of metformin from 0 to 20 mmol/L for 24, 48 or 72 h to analyze the inhibition ratio for cancer cell proliferation. *P < 0.05 versus the 0 mmol/L group. (b) MCF-7 cells were plated onto 96-well dishes and treated with EGF (50 ng/mL) and metformin (0 - 20 mmol/L) for 48 h. The survival ratio of cells was assayed by an MTT test. *P < 0.05 versus the control group. (c) Effect of EGF (50 ng/mL) on the generation of ROS in MCF-7 cells from 0 to 180 min. *P < 0.05 versus the 0 min group. (d) Effect of metformin and ROS inhibitor NAC on EGF-induced ROS generation. Cells were treated with 10 mmol/L NAC or metformin, stimulated with EGF (50 ng/mL) for 120 min, and subjected to analysis of ROS production. *P < 0.05 versus the control group; #P < 0.05 versus the EGF group. |
Metformin suppressed EGF-stimulated cell migration and invasion in MCF-7 cells
Aberrant EGF/EGFR signaling has been described as a major cause of breast cancer distant metastases [11]. Next, we cultured MCF-7 cell line in the presence of EGF to examine whether metformin could inhibit cellular migratory and invasive abilities wound-healing assay and transwell invasion assay. As shown in Figure 2a, the EGF induced-migration of MCF-7 cells was obviously inhibited by a 24 h treatment with 10 mmol/L of metformin. Metformin alone was also able to inhibit the migration of breast cancer cells without EGF treatment. To further examine the effect of metformin on the invasive ability of MCF-7 cells, we used a transwell invasion assay. The results showed that the number of cells invading to the lower chamber was increased significantly after EGF stimulation, while this EGF-induced invasive ability was terminated when treated with metformin or NAC (Fig. 2b and c). These results indicate that metformin might be an effective inhibitor of the migration and invasion of EGF-treated breast cancer cells via suppression of the ROS production.
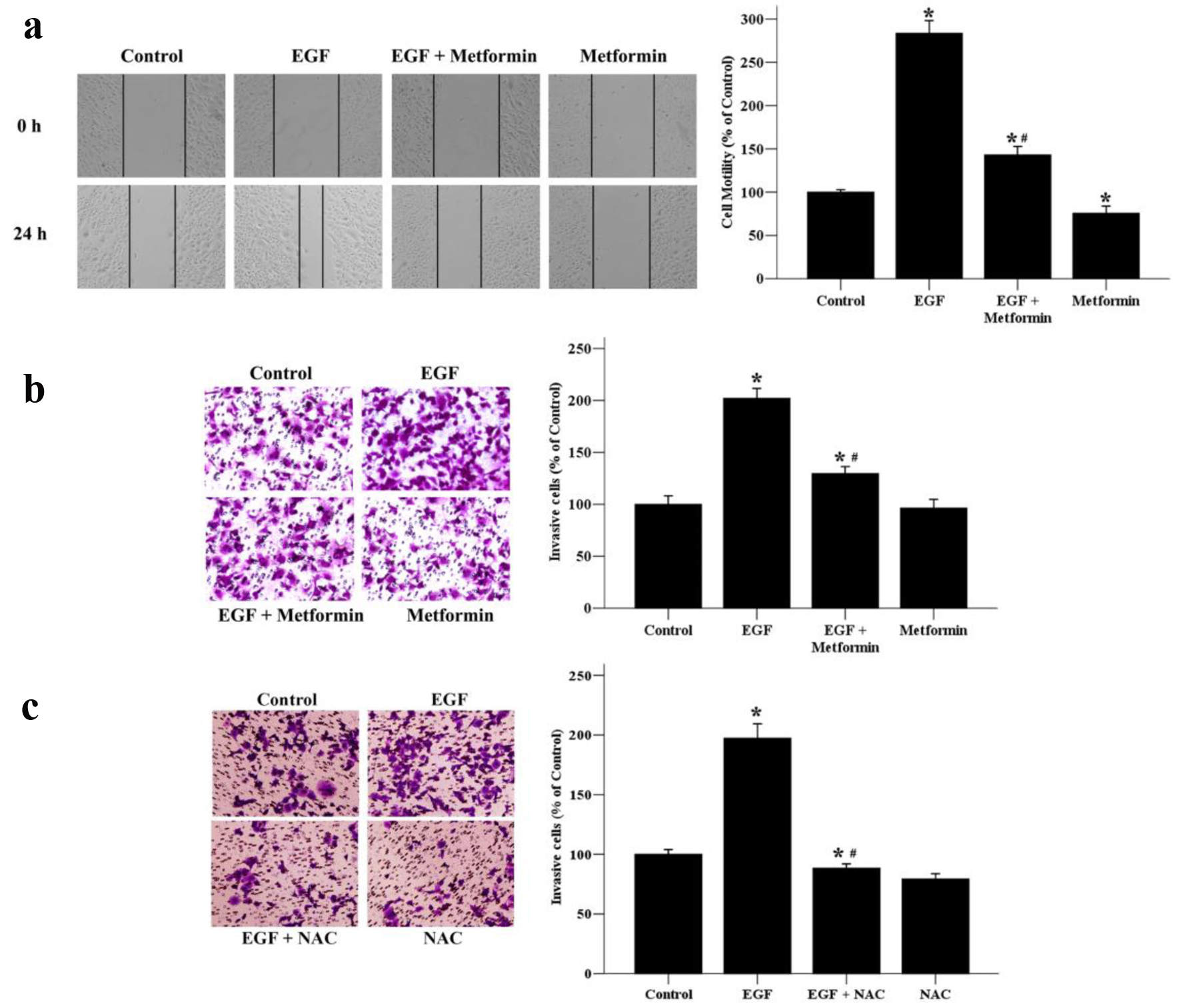 Click for large image | Figure 2. The effects of metformin on EGF-induced migratory and invasive abilities of breast cancer cells. (a) Cell migratory ability was assessed by wound healing assays. A scratch was made across confluent monolayers using a sterile pipette tip. MCF-7 cells were treated with EGF (50 ng/mL) and/or metformin (10 mmol/L) and observed after 24 h. Migrated cells were monitored using an inverted microscope with × 100 magnification. (b) A transwell invasion assay was used to quantify cell invasion. Cells were seeded into upper chambers and incubated for 48 h in the presence of EGF (50 ng/mL) and/or metformin (10 mmol/L). (c) Effect of NAC on EGF-induced cell invasion. Cells were seeded into upper chambers and incubated for 48 h in the presence of EGF (50 ng/mL) and/or NAC (10 mmol/L). The invaded cells were photographed using an inverted microscope with × 200 magnification. *P < 0.05 versus the control group. #P < 0.05 versus the EGF group. |
The inhibitory effect of metformin on EGF-induced EMT in MCF-7 cells
EMT, a transition process in which epithelial cells acquire mesenchymal characteristics, contains four important steps: 1) loss of epithelial cell adhesion; 2) gain of mesenchymal proteins; 3) degradation of surrounding basement membranes and invasion into stroma; and 4) penetrating the blood and lymph vessels and entrance to the circulation [18]. We next examined whether metformin could inhibit EGF-induced EMT, which is highly associated with cellular migratory and invasive abilities. As shown in Figure 3a, exposure of MCF-7 cells to EGF resulted in morphological alterations characteristic of EMT: after 48 h treatment with EGF, cells displayed loss of cell polarity, loss of cell-cell contact and elongation of cell shape. When EGF-stimulated MCF-7 cells were treated with metformin, an obvious morphological change from the spindle-shaped, mesenchymal form to a rounded epithelial form of cells was observed.
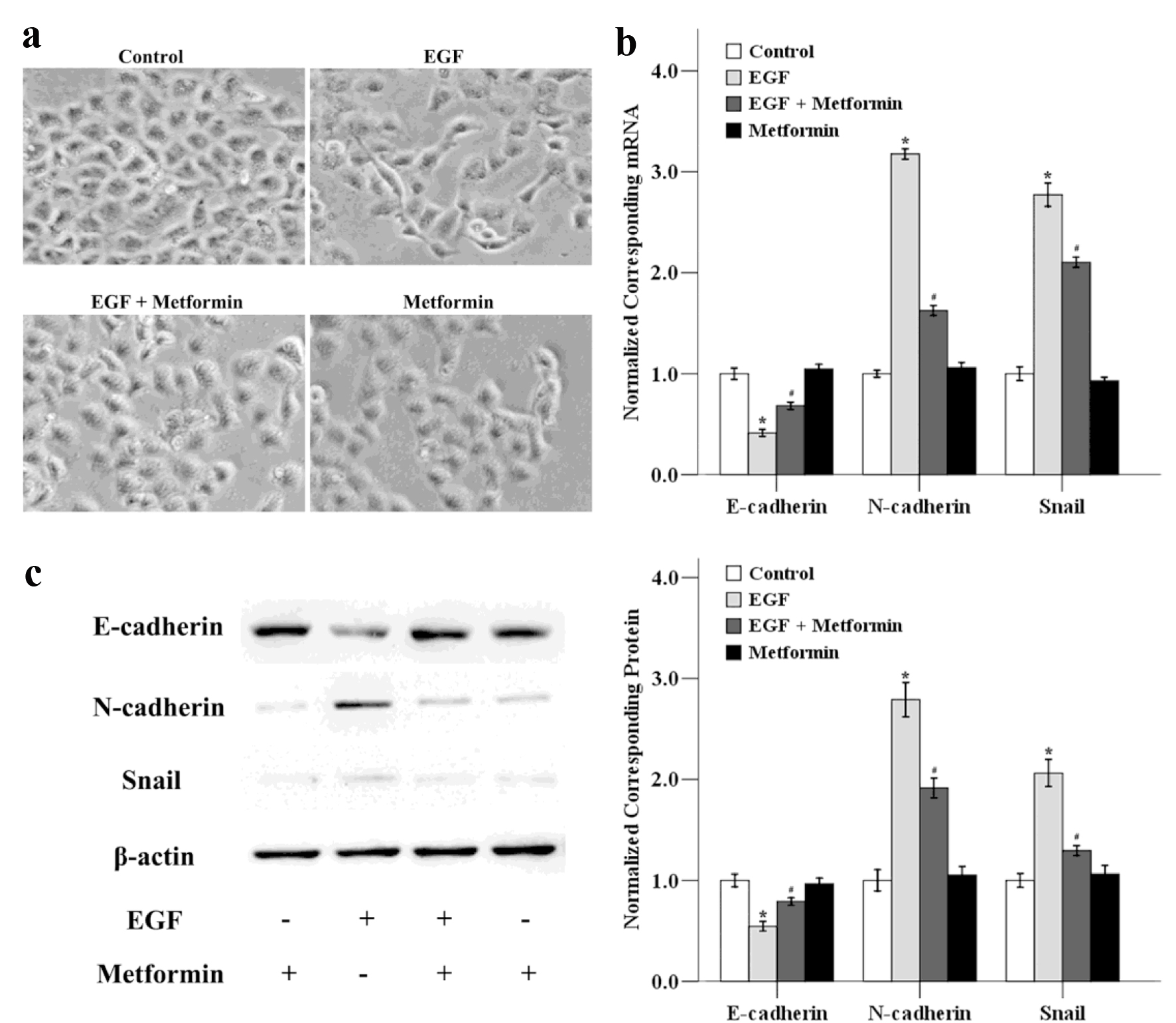 Click for large image | Figure 3. The effects of metformin on EGF-induced EMT in MCF-7 cells. (a) Cancer cells were treated with 50 ng/mL EGF and/or 10 mmol/L metformin for 48 h and the cellular morphological changes was observed. The mRNA (b) and protein (c) expression levels of E-cadherin, N-cadherin and Snail were also determined using qRT-PCR and western blotting. *P < 0.05 versus the control group; #P < 0.05 versus the EGF group. |
We next determined the expression levels of EMT-related genes after the cells were treated with EGF in the absence or presence of metformin. As illustrated in Figure 3b, EGF down-regulated the mRNA level of the epithelial marker E-cadherin, while the expression of mesenchymal markers N-cadherin and the transcriptional factor Snail were increased. Metformin could reverse these EGF-induced effects in MCF-7 cells. We also determined the expression of E-cadherin, N-cadherin and Snail at protein level using western blotting. As shown in Figure 3c, metformin could decrease the EGF-modulated EMT-related factors at the protein level, and the trend was consistent with the mRNA results. These results show that EGF-induced EMT in MCF-7 cells could be inhibited by metformin treatment.
Metformin down-regulated EGF-activated PI3K/Akt/ NF-κB pathway
Several signaling cascades involving MAPK/ERK, PI3K/Akt, and NF-κB have been reported to play key roles in EMT [8, 19]. EMT is associated with enhanced cellular invasion and motility. As shown in Figure 4, the protein levels of p-Akt and p-NF-κB were increased in MCF-7 cells with EGF treatment. We also observed that both Akt and NF-κB phosphorylation levels were strongly decreased with the addition of metformin or NAC, which indicated that metformin could inhibit EGF-induced PI3K/Akt and NF-κB signaling pathway and ROS was involved in this process. In addition, LY294002 could suppress the expression of p-NF-κB, which means that NF-κB is modulated by the Akt pathway. Taken together, our results demonstrate that metformin inhibits EGF-induced EMT which might be related with PI3K/Akt/NF-κB signaling pathway in MCF-7 cells.
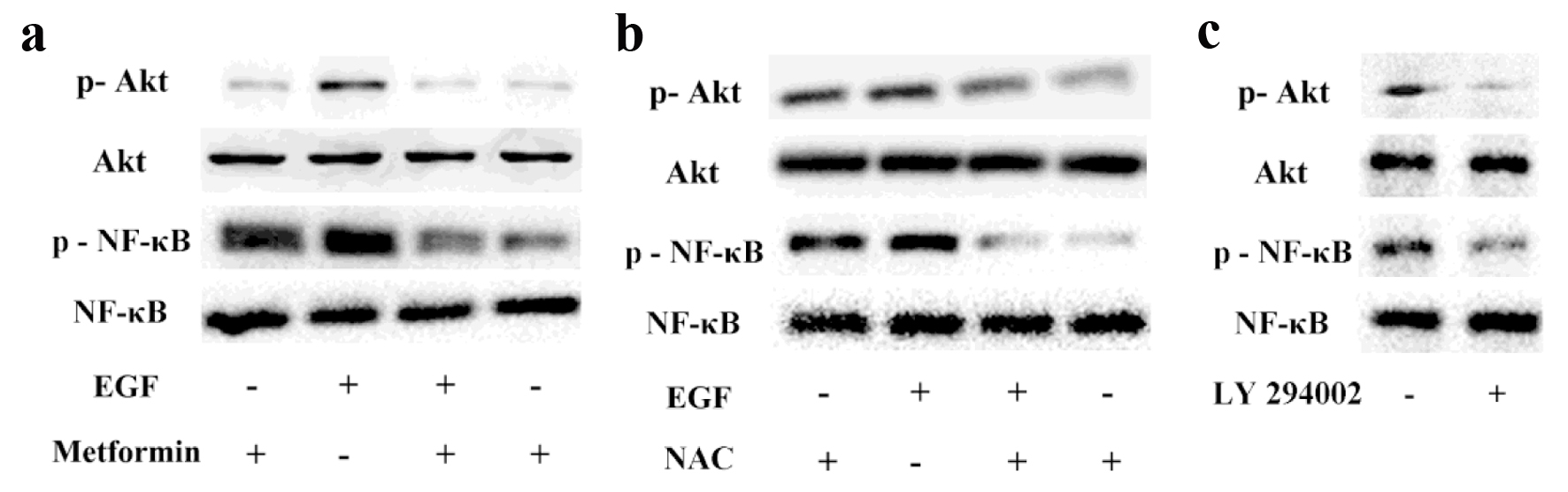 Click for large image | Figure 4. The effects of metformin on EGF-activated PI3K/Akt/NF-κB pathway. Breast cancer cells were pretreated with or without 10 mmol/L metformin (a) or NAC (b) for 24 h and then exposed to 50 ng/mL EGF to evaluate the phosphorylation levels of Akt and NF-κB via Western blotting. (c) Cancer cells were treated with LY294002 (10 µmol/L) and the activation of Akt and NF-κB were assessed via western blotting. |
The effect of the PI3K/Akt signaling pathway on EGF-induced invasion and migration in MCF-7 cells
To further assess whether the PI3K/Akt signaling pathway plays vital roles in breast cancer migration and invasion, the PI3K inhibitor LY294002 was used in the experiment. The results showed that the EGF-induced cell motility was significantly inhibited after the addition of LY294002 for 24 h (Fig. 5a). EGF-induced invasive ability was also terminated when treated with LY294002, which suggested that the PI3K/Akt signaling pathway is involved in breast cancer migration and invasion. Additionally, EGF/EGFR system has been known to be able to activate its downstream signaling pathways such as ERK and PI3K/Akt pathways [20]. In order to clarify whether EGF-induced cell migration and invasion was related with EGFR activation, we used AG 1478, an EGFR inhibitor next. Our result showed that EGFR inactivation by AG 1478 could also reverse EGF-induced cell migratory and invasive abilities. Taken together, these findings indicate that metformin could suppress EGF-induced breast cancer cell migration, invasion and EMT through the inhibition of the PI3K/Akt pathway.
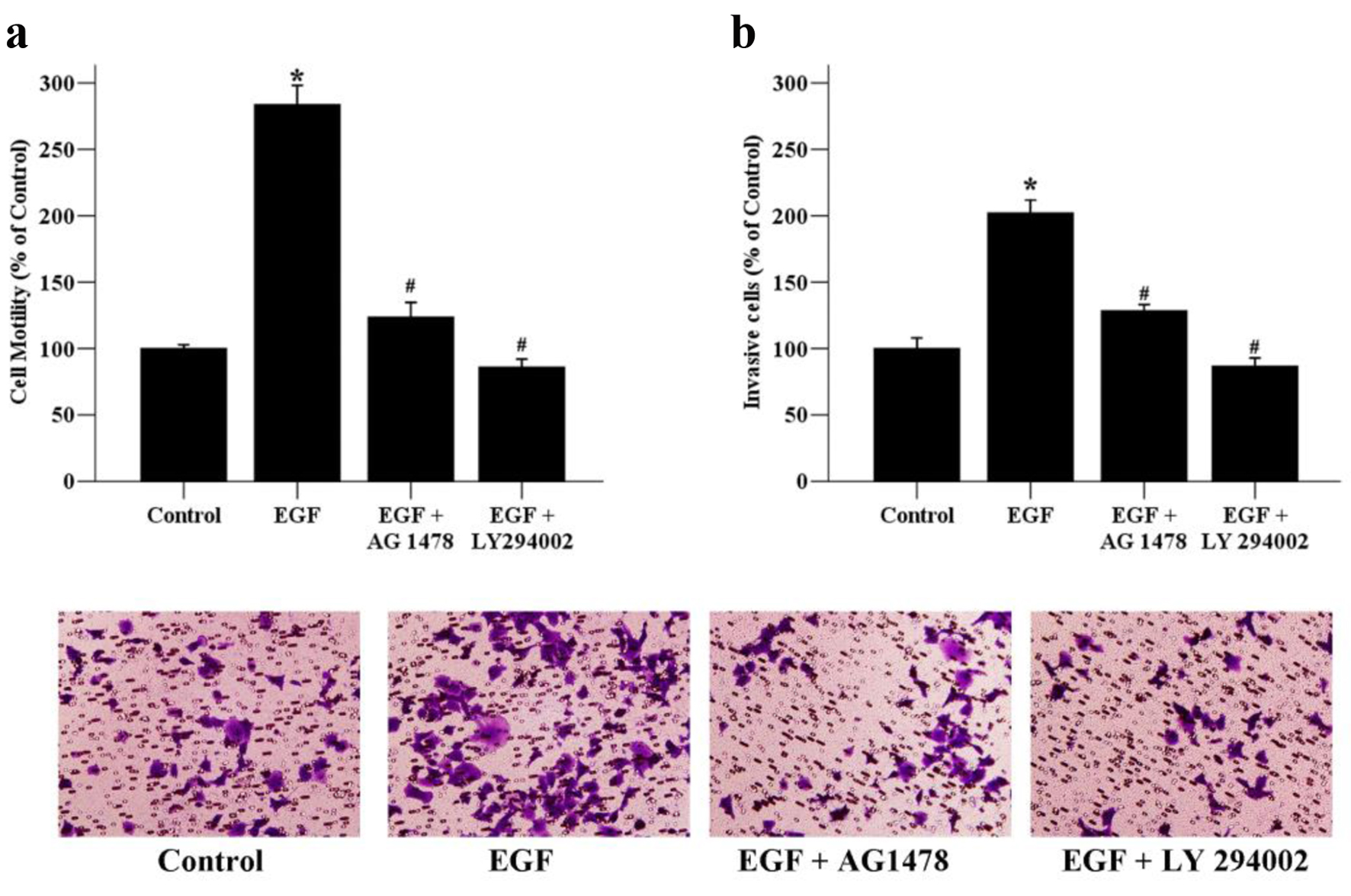 Click for large image | Figure 5. The effects of signaling inhibitors on the motility and invasion of MCF-7 cells. (a) Cancer cells were exposed to EGF (50 ng/mL) as well as AG 1478 (20 µmol/L), or LY294002 (10 µmol/L) for 24 h. The migratory ability of MCF-7 cells was assessed by wound healing assays. Migrated cells were monitored using an inverted microscope with × 100 magnification. (b) Cancer cells were exposed to EGF (50 ng/mL) as well as AG 1478 (20 µmol/L), or LY294002 (10 µmol/L) for 48 h. The invaded cells were photographed using an inverted microscope with × 200 magnification. *P < 0.05 versus the control group; #P < 0.05 versus the EGF group. |
| Discussion | ▴Top |
Breast cancer, the most frequently diagnosed tumor type and the common leading cause of cancer deaths among females worldwide, affects 1 out of 7 - 10 women [16, 21]. Multidrug resistance is the major obstacle for its treatment improvement [3]. Generally, up to 20% of elderly breast cancer patients suffer from diabetes simultaneously [22]. Metformin, a widely prescribed biguanide class oral anti-diabetic drug, has gained increasing attention because of its potential anti-tumor effects [23]. A recent study has proven that metformin can resensitize multidrug-resistant breast cancer cells, promote 5-FU-induced apoptosis, and reverse EMT phenotype of multidrug-resistant breast cancer cells [3]. In this study, we focus on whether metformin is able to suppress EGF-induced cancer proliferation invasion and EMT and its underlying mechanism.
ROS are oxygen-containing molecules that have high reactivity. Clinically, high ROS breast cancer was significantly associated with advanced cancer by AJCC stage, lymph node metastasis, and Nottingham grade [24]. In the process of EMT in malignant tumors, ROS, E-cadherin and vimentin genes play an important role. Our data showed that EGF could modulate the expression of ROS, E-cadherin and vimentin in MCF-7 cells, which further enhanced the capacity of the breast cancer MCF-7 cells to develop EMT and increase migratory and invasive ability.
Metformin was able to terminate these effects of EGF. In addition, we also tested the effects of EGF and metformin on the activation of NF-κB and Akt. Data showed that EGF significantly increased the expression levels of p-NF-κB and p-Akt in MCF-7 cells, whereas metformin resulted in an inhibition of PI3K/Akt/NF-κB pathway. Furthermore, the PI3K inhibitor, LY294002 and the EGFR inhibitor (AG 1478) could also attenuate cell migration and invasion induced by EGF.
Long-term observations and clinical trials have demonstrated decreased cancer incidence and mortality in breast cancer patients taking metformin [25]. It was reported that a 40-50% reduction in breast cancer diagnosis was observed in new metformin users of diabetic women, when followed for up to 10 years [26]. A recent meta-analysis showed that the use of metformin in diabetic patients diagnosed with cancer was associated with a decrease in risk of breast cancer (RR = 0.70) [27]. Our previous retrospective clinical study showed that metformin is associated with luminal breast cancer and can inhibit the invasion and metastasis of breast cancer [28]. Many in vitro and in vivo studies using mouse xenograft models have proven that metformin exerts anti-tumor activity via inhibition of cancer cell proliferation, promotion of cancer cell apoptosis, suppression of tumor cell migration and invasion, decreasing of tumor angiogenesis, cooperating with chemotherapeutic drugs as well as immunomodulation [29-32]. Queiroz et al [31] recently showed that metformin could suppress the proliferation of breast cancer MCF-7 cells by promoting cell cycle arrest in the G0-G1 phase, inhibiting cyclin D1 and leading to cell apoptosis and cell death, which were associated with oxidative stress, AMPK and FOXO3a activation. Liu et al [32] proved that metformin inhibited triple negative breast cancer cell proliferation via S phase cell cycle arrest. Angiogenesis is a fundamental event in the process of tumor growth [33]. Metformin treatment has also been proven to decrease tumor angiogenesis via targeting of epidermal growth factor receptor-2 (HER2)/hypoxia inducible factor 1α (HIF-1α)/vascular endothelial growth factor (VEGF) secretion axis [29]. In addition, metformin potentiates the immune system by inhibiting immune exhaustion of CD8+ tumor-induced lymphocytes, thus enhancing the immune response mediated by T cell to cancer cells [30]. In the current study, we found that metformin could inhibit EGF-induced breast cancer cell proliferation, invasion and migration.
EMT, an important phenomenon that governs metastasis during cancer progression, is a transition process in which epithelial cells acquire mesenchymal characteristics. The hallmarker of the EMT is the loss of cellular polarity and epithelial markers (E-cadherin) and the acquisition of mesenchymal markers (vimentin and N-cadherin) and invasive property as well as the up-regulation of associated transcriptional regulators (Snail, Twist) [6]. EMT facilitates tumor migration from the origin site and dissemination to distant tissues. EMT is also associated with the evasion of apoptosis and senescence and anoikis resistance in breast cancer cells [34, 35]. The stimulation of cancer cells with various growth factors, such as transforming growth factor beta (TGF-β) and EGF, can induce EMT and ultimately increase cell migration and metastasis [8, 20]. EGF/EGFR system is implicated in regulating mammary gland morphogenesis and development, while aberrant EGF/EGFR system and its downstream signaling pathways including PI3K/Akt and ERK pathways are associated with breast cancer progression [9, 20]. The activation of Rac1 and generation of ROS might be involved in EGF-induced tumor invasion [36]. Our study also proved that EGF was able to promote the development of EMT in breast cancer cells, which could be terminated by metformin. Other study shows that high EGFR tumors attract immune cells (anti-cancer immune cells) and are associated with high expression of immune checkpoint molecules, especially the ER-positive/HER2-negative breast cancer subtype [37]. Therefore, we use EGFR monoclonal antibody to treat metastatic breast cancer with poor efficacy, while we inhibit the downstream signaling of EGF/EGFR pathway and have better efficacy.
Metformin has been proven to exert its anti-cancer action via AMPK-dependent and/or AMPK-independent mechanisms. AMPK, a highly conserved serine/threonine heterotrimeric protein kinase consisting of one catalytic (α) and two regulatory (β and γ) subunits, plays an important role in regulating metabolic pathways, such as gluconeogenesis, protein and fatty acid synthesis [38]. In cancer cells, the stimulation of AMPK by metformin resulted in the inhibition of tumor growth and invasion [31]. AMPK is linked with the PI3K/Akt and MAPK/extracellular signal-regulated kinases (ERK) cascades [16]. Banerjee et al [39] recently showed that metformin could attenuate ERK signaling by activating AMPK pathway leading to the suppression of transcriptional factors Snail and Slug, which further result in the upregulation of E-cadherin. Chou et al [40] also proved that AMPK plays an important role in regulating EMT in breast cancer cells by upregulating Foxo3a signaling through an Akt-dependent mechanism. Our result showed that metformin could inhibit EGF-induced EMT in MCF-7 cell via PI3K/Akt/NF-κB pathway.
In conclusion, the current study demonstrates that metformin plays an important role in inhibiting EGF-induced proliferation, migration, invasion and EMT of breast cancer cells in vitro by blocking the PI3K/Akt/NF-κB pathway. These results suggest that metformin might be a potential anti-cancer agent for the treatment of breast cancer.
Acknowledgments
None to declare.
Financial Disclosure
This study was supported by grants from the Key Project of Shaanxi International Science and Technology Cooperation and Exchange Program (2014KW23-07, 2019KW-35, 2021KW-67), Free Exploration Project of the Second Affiliated Hospital of Xi’an Jiaotong University (No. 2020YJ (ZYTS)611), and Medical Technology Access Program of the Second Affiliated Hospital of Xi’an Jiaotong University (No. 2018(56)).
Conflict of Interest
The authors have declared that no competing interest exists.
Informed Consent
Not applicable.
Author Contributions
MW, WB and ZS performed the experiments. LB, ZL, LS and ZY designed and supervised the study. MW, HY and MQ were involved with data analysis and wrote the manuscript. All authors read and approved the final manuscript.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
| References | ▴Top |
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7-30.
doi pubmed - Weigelt B, Peterse JL, van 't Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5(8):591-602.
doi pubmed - Qu C, Zhang W, Zheng G, Zhang Z, Yin J, He Z. Metformin reverses multidrug resistance and epithelial-mesenchymal transition (EMT) via activating AMP-activated protein kinase (AMPK) in human breast cancer cells. Mol Cell Biochem. 2014;386(1-2):63-71.
doi pubmed - Felipe Lima J, Nofech-Mozes S, Bayani J, Bartlett JM. EMT in breast carcinoma-a review. J Clin Med. 2016;5(7):65.
doi pubmed - Wang Y, Shang Y. Epigenetic control of epithelial-to-mesenchymal transition and cancer metastasis. Exp Cell Res. 2013;319(2):160-169.
doi pubmed - Wendt MK, Smith JA, Schiemann WP. Transforming growth factor-beta-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene. 2010;29(49):6485-6498.
doi pubmed - Kim J, Kong J, Chang H, Kim H, Kim A. EGF induces epithelial-mesenchymal transition through phospho-Smad2/3-Snail signaling pathway in breast cancer cells. Oncotarget. 2016;7(51):85021-85032.
doi pubmed - Li L, Qi L, Liang Z, Song W, Liu Y, Wang Y, Sun B, et al. Transforming growth factor-beta1 induces EMT by the transactivation of epidermal growth factor signaling through HA/CD44 in lung and breast cancer cells. Int J Mol Med. 2015;36(1):113-122.
doi pubmed - Hardy KM, Booth BW, Hendrix MJ, Salomon DS, Strizzi L. ErbB/EGF signaling and EMT in mammary development and breast cancer. J Mammary Gland Biol Neoplasia. 2010;15(2):191-199.
doi pubmed - Davis FM, Peters AA, Grice DM, Cabot PJ, Parat MO, Roberts-Thomson SJ, Monteith GR. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS One. 2012;7(5):e36923.
doi pubmed - Giltnane JM, Moeder CB, Camp RL, Rimm DL. Quantitative multiplexed analysis of ErbB family coexpression for primary breast cancer prognosis in a large retrospective cohort. Cancer. 2009;115(11):2400-2409.
doi pubmed - Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160-1174.
doi pubmed - Grossmann ME, Yang DQ, Guo Z, Potter DA, Cleary MP. Metformin treatment for the prevention and/or treatment of breast/mammary tumorigenesis. Curr Pharmacol Rep. 2015;1(5):312-323.
doi pubmed - Wahdan-Alaswad R, Harrell JC, Fan Z, Edgerton SM, Liu B, Thor AD. Metformin attenuates transforming growth factor beta (TGF-beta) mediated oncogenesis in mesenchymal stem-like/claudin-low triple negative breast cancer. Cell Cycle. 2016;15(8):1046-1059.
doi pubmed - Zhang Y, Storr SJ, Johnson K, Green AR, Rakha EA, Ellis IO, Morgan DA, et al. Involvement of metformin and AMPK in the radioresponse and prognosis of luminal versus basal-like breast cancer treated with radiotherapy. Oncotarget. 2014;5(24):12936-12949.
doi pubmed - Liu H, Scholz C, Zang C, Schefe JH, Habbel P, Regierer AC, Schulz CO, et al. Metformin and the mTOR inhibitor everolimus (RAD001) sensitize breast cancer cells to the cytotoxic effect of chemotherapeutic drugs in vitro. Anticancer Res. 2012;32(5):1627-1637.
- Kozlova N, Samoylenko A, Drobot L, Kietzmann T. Urokinase is a negative modulator of Egf-dependent proliferation and motility in the two breast cancer cell lines MCF-7 and MDA-MB-231. Mol Carcinog. 2016;55(2):170-181.
doi pubmed - Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871-890.
doi pubmed - Vergara D, Valente CM, Tinelli A, Siciliano C, Lorusso V, Acierno R, Giovinazzo G, et al. Resveratrol inhibits the epidermal growth factor-induced epithelial mesenchymal transition in MCF-7 cells. Cancer Lett. 2011;310(1):1-8.
doi pubmed - Tsai PC, Fu YS, Chang LS, Lin SR. Taiwan cobra cardiotoxin III suppresses EGF/EGFR-mediated epithelial-to-mesenchymal transition and invasion of human breast cancer MDA-MB-231 cells. Toxicon. 2016;111:108-120.
doi pubmed - Anderson GF, Chu E. Expanding priorities—confronting chronic disease in countries with low income. N Engl J Med. 2007;356(3):209-211.
doi pubmed - Wolf I, Sadetzki S, Catane R, Karasik A, Kaufman B. Diabetes mellitus and breast cancer. Lancet Oncol. 2005;6(2):103-111.
doi - Quinn BJ, Kitagawa H, Memmott RM, Gills JJ, Dennis PA. Repositioning metformin for cancer prevention and treatment. Trends Endocrinol Metab. 2013;24(9):469-480.
doi pubmed - Oshi M, Gandhi S, Yan L, Tokumaru Y, Wu R, Yamada A, Matsuyama R, et al. Abundance of reactive oxygen species (ROS) is associated with tumor aggressiveness, immune response, and worse survival in breast cancer. Breast Cancer Res Treat. 2022;194(2):231-241.
doi pubmed - Chae YK, Arya A, Malecek MK, Shin DS, Carneiro B, Chandra S, Kaplan J, et al. Repurposing metformin for cancer treatment: current clinical studies. Oncotarget. 2016;7(26):40767-40780.
doi pubmed - Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32(9):1620-1625.
doi pubmed - Zhang ZJ, Li S. The prognostic value of metformin for cancer patients with concurrent diabetes: a systematic review and meta-analysis. Diabetes Obes Metab. 2014;16(8):707-710.
doi pubmed - Min W, Wang B, Guo A, Mao G, Zhao Y, Zhang S, He R, et al. The effect of metformin on the clinicopathological features of breast cancer with type 2 diabetes. World J Oncol. 2020;11(1):23-32.
doi pubmed - Wang J, Li G, Wang Y, Tang S, Sun X, Feng X, Li Y, et al. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF-1alpha/VEGF secretion axis. Oncotarget. 2015;6(42):44579-44592.
doi pubmed - Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci U S A. 2015;112(6):1809-1814.
doi pubmed - Queiroz EA, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ, Barbosa AM, et al. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One. 2014;9(5):e98207.
doi pubmed - Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle. 2011;10(17):2959-2966.
doi pubmed - Oshi M, Newman S, Tokumaru Y, Yan L, Matsuyama R, Endo I, Nagahashi M, et al. Intra-tumoral angiogenesis is associated with inflammation, immune reaction and metastatic recurrence in breast cancer. Int J Mol Sci. 2020;21(18):6708.
doi pubmed - Davis FM, Azimi I, Faville RA, Peters AA, Jalink K, Putney JW, Jr., Goodhill GJ, et al. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene. 2014;33(18):2307-2316.
doi pubmed - Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704-715.
doi pubmed - Yang Y, Du J, Hu Z, Liu J, Tian Y, Zhu Y, Wang L, et al. Activation of Rac1-PI3K/Akt is required for epidermal growth factor-induced PAK1 activation and cell migration in MDA-MB-231 breast cancer cells. J Biomed Res. 2011;25(4):237-245.
doi - Oshi M, Gandhi S, Tokumaru Y, Yan L, Yamada A, Matsuyama R, Ishikawa T, et al. Conflicting roles of EGFR expression by subtypes in breast cancer. Am J Cancer Res. 2021;11(10):5094-5110.
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8(10):774-785.
doi pubmed - Banerjee P, Surendran H, Chowdhury DR, Prabhakar K, Pal R. Metformin mediated reversal of epithelial to mesenchymal transition is triggered by epigenetic changes in E-cadherin promoter. J Mol Med (Berl). 2016;94(12):1397-1409.
doi pubmed - Chou CC, Lee KH, Lai IL, Wang D, Mo X, Kulp SK, Shapiro CL, et al. AMPK reverses the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis. Cancer Res. 2014;74(17):4783-4795.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.