

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Review
Volume 13, Number 6, December 2022, pages 337-342

Primary Angiosarcoma of the Spleen: An Aggressive Neoplasm
Mira Damounya, Subhi Mansoura, Safi Khuria, b, c
aGeneral Surgery Department, Rambam Medical Center, Haifa, Israel
bHPB and Surgical Oncology Unit, Rambam Medical Center, Haifa, Israel
cCorresponding Author: Safi Khuri, HepatoPancreatoBiliary Surgical Unit, General Surgery Department, Rambam Medical Care Center, Haa’leya Hashniya, Haifa 31096, Israel
Manuscript submitted October 31, 2022, accepted November 23, 2022, published online December 1, 2022
Short title: Angiosarcoma of the Spleen
doi: https://doi.org/10.14740/wjon1542
- Abstract
- Introduction
- Methods
- Demographic Features
- Clinical Presentation
- Diagnosis
- Management
- Summary
- References
| Abstract | ▴Top |
Primary tumors of the spleen are rare, with an incidence rate of about 0.1%. These tumors could be benign, usually asymptomatic, or malignant which are usually symptomatic with abdominal pain being the most common symptom. Lymphoid neoplasms are the most common primary splenic tumors. Primary angiosarcoma is one of the extremely rare malignant vascular neoplasms of the spleen, which carries a dismal prognosis. It constitutes almost 7.4% of all primary malignant splenic neoplasms and is well known as an aggressive tumor with high local recurrence and distant metastasis rates. Overall survival is up to 12 months following diagnosis, regardless of management strategy. Due to the broad differential diagnosis of splenic tumors, this tumor is often forgotten, and is very challenging to diagnose early. Less than 300 cases of primary splenic angiosarcoma have been reported in the English literature. The main issue of this article is to review the current English literature to figure out the characteristic demographic features, clinical presentation, imaging findings and management of such tumors, in order to increase awareness of the treating physicians to improve diagnosis, management, as well as overall survival.
Keywords: Angiosarcoma of the spleen; Rare tumor; Dismal prognosis
| Introduction | ▴Top |
Tumors of the spleen are usually rare and can be classified as primary or secondary, with the latter being less common and develops as a metastasis from a remote source, such as carcinoma of the colon, ovary, breast, lung, and melanoma. Primary splenic tumors are extremely rare, with an overall incidence rate of almost 0.1% [1]. Different classification systems exist for these tumors, benign versus malignant or lymphoid versus non-lymphoid tumors. Benign splenic tumors, such as hamartoma, hemangioma or lymphangioma, are usually asymptomatic and are discovered incidentally by radiological exams done for unrelated indications [2]. These tumors become symptomatic when they reach a large size, causing direct pressure on nearby organs (e.g., early satiety due to pressure on the stomach or abdominal pain), and to a lesser extent, spontaneous rupture of the spleen [2]. On the other hand, primary malignant tumors are usually symptomatic, with abdominal pain being the most common presentation [3]. Lymphoid tumors are the most common primary tumors of the spleen [4], and include Hodgkin’s disease, non-Hodgkin lymphoma, plasmacytoma, Castleman’s tumor and others. Non-lymphoid primary malignant splenic tumors, such as fibrosarcoma, leiomyosarcoma, Kaposi’s sarcoma, malignant teratoma and angiosarcoma, are very uncommon tumors [1].
Primary angiosarcoma of the spleen, also known as hemangiosarcoma and firstly described by Langhans in 1879 [3], is an extremely rare primary malignant vascular tumor arising from vascular endothelium of the spleen specifically mesenchymal-derived elongated endothelial cells lining the network of splenic sinusoids [5]. Less than 300 cases of primary angiosarcoma of the spleen were reported in the English literature. This tumor is very aggressive and carries a dismal prognosis, even when treated accordingly and in a timely manner. The dismal prognosis for such tumor is mainly related to its biologic features: highly infiltrated with anaplastic cells which are rapidly proliferating, and thus, high local recurrence and systemic metastasis rates [6]. The overall survival for the majority of patients is up to 12 months following diagnosis (median of 4 - 18 months), regardless of treatment strategy [3, 7]. It is well known as a fatal tumor of unknown pathogenesis and constitutes less than 1% and 7.4% of all sarcoma tumors and all primary malignant splenic tumors, respectively [8, 9]. The estimated annual incidence rate for primary splenic angiosarcoma is almost one case per 4 million patients [3, 9]. In light of broad differential diagnosis, which may include other benign vascular tumors and malignant nonvascular tumors, this tumor is often forgotten, and thus, challenging to diagnose early.
The main issue of this article is to review the current English literature to grasp the characteristic demographic features, clinical presentation, radiological findings, and management of such tumors. This is in order to increase awareness of the treating physician to improve diagnosis, management, consequently improving overall survival.
| Methods | ▴Top |
A search in PubMed was conducted, based on the “PICOS” acronym. Headings and text words were used to identify studies (in the form of case reports or case series) published discussing angiosarcoma of the spleen.
The following search terms were included: “spleen tumors”, “splenic angiosarcoma”, “primary splenic malignancy”, “angiosarcoma”, and “rare tumors of the spleen”.
Initial search of the current literature revealed almost 300 cases of primary angiosarcoma of the spleen. Most of the reported cases were in the form of case reports or case series.
Extracted data included demographic features (patients’ age, gender, risk factors), clinical presentation, radiological findings by either abdominal ultrasound (US) scan, abdominal computed tomography (CT) scan or abdominal magnetic resonance imaging (MRI), management (surgical and oncological) and follow-up.
| Demographic Features | ▴Top |
The precise incidence rate of primary angiosarcoma of the spleen is unknown, yet it is believed to be 0.14 - 0.23 cases/1 million patient [10]. It is even rarer in the pediatric age group (less than 18 years old), with very few cases reported (up to 10 reported cases) in the English literature [11]. It can develop at any age group with a wide age range (14 month to 89 years old). The mean age at diagnosis differs between males and females. Females usually presents at an older age (68 years old) than males (53 years old) [9]. The mean age at diagnosis for both sexes is 59 years old (usually develops during the fifth to sixth decade of life) [10]. Although there is no apparent gender predilection, it is slightly more common in males than females. Male to female (M/F) ratios of 1.1:1, 1.3:1, and 1.4:1 were reported in different case series [3, 9, 12]. The precise pathogenesis for the development of this tumor is unknown. Several environmental factors (such as monomer vinyl chloride and thorium dioxide) were raised as risk factors for angiosarcoma of the spleen. In addition, as few cases of splenic angiosarcoma developed years following radiotherapy for other malignancies, chemo/radiotherapeutic agents were also proposed as risk factors as well [12]. Up to date, and in contrast to the direct relationship between the aforementioned risk factors and liver angiosarcoma, there is no scientific based correlation between the previously mentioned factors and splenic angiosarcoma development [13]. The previous hypothesis is based on small series of reports and case reports; therefore, future studies are required to elucidate this correlation.
| Clinical Presentation | ▴Top |
Primary splenic angiosarcoma is usually symptomatic on diagnosis [1]. Patients usually present with nonspecific symptoms, which increase difficulty for early diagnosis. A wide range of clinical presentation (mild abdominal pain to hemodynamic instability due to spontaneous rupture) has been reported. The most common presenting symptom is abdominal pain reported in 75-83% of cases [4, 9]. In his study which included 12 patients, Thompson et al [13] documented abdominal pain as the most common presentation in 67% of patients. The pain can be described as a mild upper abdominal pain, when it is due to splenomegaly, or severe sharp pain in the case of splenic rupture. Other less common symptoms include fatigue, weight loss, fever and chest pain being reported in 5-39%, 10-40%, 10% and 10%, respectively [3, 9]. An extremely rare clinical presentation mentioned in case reports include upper gastrointestinal bleeding (due to bleeding ulcer), hemoptysis, loss of appetite, and shortness of breath (Table 1). Spontaneous rupture of the spleen with hemoperitoneum is a clinical emergency, which fortunately occurs only in up to 32% of patients with primary angiosarcoma of the spleen [9]. Rupture of the splenic tumor leads to early metastasis to other intra-abdominal organs; hence, it is known as the worst prognostic factor [14]. Worth mentioning, in contrast to the slight male predominance reported earlier, most patients who presented with spontaneous splenic rupture due to primary angiosarcoma were females, with F/M ratio of 1.6:1 [15-33]. This surgical emergency may affect a wide age range of patients, with the youngest patient being 17 years old female [17], while the oldest was a female in her 80s, reported by Sivelli et al [25]. The most common clinical presentation following spontaneous rupture of the spleen was acute upper abdominal pain reported by all patients. Hemodynamic instability was also a common presentation as well, documented in 68% of patients with spontaneous rupture [15, 17, 19, 21, 23-27, 29-31, 33]. In statistical terms, hemodynamic instability may develop in up to 21% of patients with angiosarcoma of the spleen. Same admission mortality rate for patients presenting with spontaneous rupture and shock was 27% (Table 2). There was an obvious direct relationship between diameter and weight of the spleen and risk of spontaneous rupture, as most reported cases demonstrated an enlarged overweight spleen (range of diameter: 12 - 31 cm (normal: 12 cm)/range of weight: 800 - 5,200 g (normal: 28 - 226 g)).
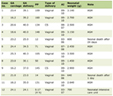 Click to view | Table 1. The Clinical Presentation for Patients With Splenic Angiosarcoma |
Click to view | Table 2. Reported Cases of Spontaneous Spleen Rupture Due to Primary Angiosarcoma |
| Diagnosis | ▴Top |
Being an extremely rare tumor, in addition to presenting with nonspecific symptoms, diagnosis of primary angiosarcoma of the spleen is a challenging issue to the treating physician. The diagnosis of such tumors entails a constellation of physical, radiological, and histopathological findings.
Physical examination of patients with primary angiosarcoma of the spleen can be without abnormal findings [34], while on the other hand abdominal distention, due to hemoperitoneum in the case of rupture, could be found. As most patients usually exhibit a replacement of the normal splenic parenchyma by tumor cells, diffuse enlargement of the entire spleen with splenomegaly (77-94%) is the most common physical finding [3, 5]. Solitary splenic masses, although reported, are less common [6]. In his study, Neuhauser et al [9] reported palpable spleen due to splenomegaly as the most common physical finding, shown in 71% (20/28 patients). Hepatomegaly, due to metastatic lesions, was noticed in only four patients. In other study [13], the most common clinical finding, demonstrated in almost 60% of patients, was a complex mass or masses in an enlarged spleen. As have been mentioned before, a life-threatening surgical emergency may be the first presentation in case of spontaneous rupture of the primary tumor and hemodynamic instability.
In spite of the significant technological progress in imaging techniques during the recent years, radiological diagnosis of primary splenic angiosarcoma is still very challenging [6]. The difficulty arises mainly due to the radiological overlap between this tumor and other vascular (hemangiomas, epithelioid vascular tumors, and littoral cell angiomas) and nonvascular (lymphomas) tumors of the spleen [13]. Nowadays, several radiological tests are available for the diagnosis and staging workup purposes for splenic angiosarcoma. These include abdominal US, CT scan and MRI. The most common radiological finding, seen in all the previously mentioned techniques, is splenomegaly [6].
Heterogeneous complex mass, including areas of necrosis and hemorrhage, is the most common US finding [7]. The necrotic and the hemorrhagic areas are usually demonstrated as cystic areas within the splenic mass.
CT scan is another modality used for diagnosis as well as for staging of splenic angiosarcoma. Findings on CT scan depend usually on whether the primary tumor ruptured. In the presence of an unruptured tumor, CT scan findings are similar to the previously mentioned findings by US: heterogeneous splenic mass with indistinct borders. On the other hand, in the setting of tumor rupture, hyperattenuation, due to bleeding, on unenhanced images will be prominent [13]. The most common organs to be involved by metastasis from splenic angiosarcoma are the liver (the most common), lungs, bones, lymph nodes, gastrointestinal tract, brain, and adrenal glands. As have been mentioned before, CT scan is a useful tool for staging and the detection of remote metastasis to other organ systems. Secondary metastatic lesions usually appear hypervascular on CT scan [9].
In abdominal MRI, areas of increased and decreased signal intensity are noticed on T1 and T2 weighted pulse sequences. These signals are consistent with the presence of necrosis and hemorrhage within the tumor [7].
The precise diagnosis of primary angiosarcoma of the spleen is usually made by histopathological and immunohistochemical tests.
Macroscopically, solitary/isolated masses are uncommon, while diffuse splenic parenchyma involvement by tumor cells is the norm [6]. Microscopically, splenic angiosarcoma contains poorly differentiated regions consisting of sarcomatous features, and well differentiated areas made of sinuses similar to splenic sinuses [6]. Disorganized vascular channels lined by atypical large endothelial cells with irregular nuclei constitute the tumor mass. Immunohistochemical tests have proved that focal vasoformative component is the dominant pattern for splenic angiosarcomas [9, 11]. Poorly formed lamina lined by endothelial cells highlighted by vascular markers CD31 and factor VIII help in establishing the diagnosis of splenic angiosarcoma. Less common vascular markers, which could stain positive by immunohistochemical tests include CD34, vimentin and ETS-related gene (ERG).
In contrast to other sarcoma subtypes, tumor grade and histological appearance are not considered prognostic factors in patients with primary splenic angiosarcoma, thus, the previously mentioned factors are not related to tumor outcomes [35]. On the other hand, tumor size and mitotic rate were found to be prognostic factors in other study [36], which included 55 patients with angiosarcoma.
| Management | ▴Top |
Due to the small number of reported cases in the English literature, in addition to the absence of studies (retrospective/prospective), treatment of patients with primary angiosarcoma of the spleen is not scientific based. Each case is treated separately based on physician experience. Thus, it is highly advisable to treat this group of patients with a multidisciplinary team of physicians, composed of qualified surgeons, specialized oncologist, and radiologists.
Treatment usually includes a combination of splenectomy, radiotherapy, and chemotherapy, although this does not affect the median overall survival, which is 5 - 6 months [1].
Although rarely curative, surgical resection (splenectomy) is considered the mainstay of management, especially in the presence of local disease confined to the spleen or spontaneous rupture [3, 9, 12]. As previously mentioned, rupture is the worst prognostic factor for splenic angiosarcoma. In their study, Montemayor et al [37] demonstrated a mean survival rate following splenectomy without rupture of 14.4 months, with a 4.4-month mean survival following rupture. In his article, Hsu et al [11] reported the longest disease-free survival (16 years) following splenectomy due to splenic angiosarcoma without adjuvant chemo/radiotherapy.
Splenic angiosarcoma is highly refractory to chemo/radiotherapy treatment, and up to date, there is no scientific evidence to suggest these treatment modalities for such tumors [9, 12]. Some recent reported cases are exceptional to the previously mentioned statement: Wheelwright et al [34] reported two cases of splenic angiosarcoma treated by surgical means and adjuvant chemotherapy. One patient, treated by adjuvant doxorubicin and ifosfamide-based chemotherapy, showed no evidence of recurrence at 4 years of clinical and radiological follow-up. The second patient, also treated with adjuvant doxorubicin and ifosfamide-based chemotherapy, developed bone metastasis at 3 years’ follow-up, with a stable disease at 5 years’ clinical and radiological follow-up. In another reported case [6], the patient showed progressive disease under adjuvant gemcitabine-based chemotherapy, and stable disease following a second-line doxorubicin and ifosfamide-based chemotherapy. These findings suggest that adjuvant doxorubicin-based chemotherapy is promising. Yet, physicians should be careful as it is based on single case report and future studies are required to examine the effectiveness of such regimens.
Prognosis of primary angiosarcoma of the spleen is very poor. Most patient (69-100%) develop metastatic disease, with the liver (89%) being the most affected organ, followed by the lungs (78%), lymphatic system (56%), and skeletal system (22%) [38].
| Summary | ▴Top |
Primary angiosarcoma of the spleen is an extremely rare aggressive neoplasm, which carries a dismal prognosis due to its biological features and late diagnosis [6]. It encompasses several medical fields in its realm. This rare tumor with its puzzling clinical manifestations carries a hefty diagnostic challenge to the treating team of physicians. The main difficulty is the early diagnosis and the differential diagnosis between the aggressive angiosarcoma tumor that carries a dismal prognosis and the other splenic primary tumors which carry a better prognosis, when diagnosed early and treated accordingly. With the paucity and relative weakness of available data in the current English literature, one needs to carefully review all parameters in different aspects, including demographic features, clinical presentation, imaging tests, and histopathological findings. Physicians must have a low threshold of suspicion to try and prevent missing such an aggressive tumor.
Spontaneous rupture of the tumor, a surgical emergency more common in female patients with splenic angiosarcoma, may develop in up to 30% of patients [9]. Mortality rate following tumor rupture may be as high as 27%. Rupture is the worst prognostic factor. The most obvious physical finding is a palpable spleen (due to splenomegaly)/abdominal mass at the left upper quadrant of the abdomen [3, 5]. Enlargement of the spleen along with heterogeneous complex mass, are the most common radiological findings [7, 13]. Macroscopic and microscopic findings are very characteristics and include diffuse involvement of the spleen and disorganized vascular channels lined with atypical large endothelial cells with irregular nuclei, respectively. Vascular markers including CD31 and factor VIII are useful for diagnosis establishment. Controversy exists regarding the best therapeutic approach, and a multidisciplinary team of physicians is highly advisable. Treatment usually includes surgical (splenectomy) and oncological (chemotherapy/radiotherapy) modalities. Future studies are needed, preferably multi-centric and prospective in nature, in order to improve the level of evidence and improve our understanding of the disease process, diagnosis, and treatment.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The authors have no conflict of interest to declare.
Author Contributions
Acquisition of the search was made by Mira Damouny and Subhi Mansour. The paper was drafted by Subhi Mansour and Safi Khuri. Critical revision and final approval of the published version were done by Safi Khuri.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Fotiadis C, Georgopoulos I, Stoidis C, Patapis P. Primary tumors of the spleen. Int J Biomed Sci. 2009;5(2):85-91.
- Willcox TM, Speer RW, Schlinkert RT, Sarr MG. Hemangioma of the spleen: presentation, diagnosis, and management. J Gastrointest Surg. 2000;4(6):611-613.
doi pubmed - Falk S, Krishnan J, Meis JM. Primary angiosarcoma of the spleen. A clinicopathologic study of 40 cases. Am J Surg Pathol. 1993;17(10):959-970.
doi pubmed - Langhanis T. Pulsating cavernous neoplasm of the spleen with metastatic nodules to the liver. Virchows Arch (Pathol Anat). 1879;75:273-291.
- Hai SA, Genato R, Gressel I, Khan P. Primary splenic angiosarcoma: case report and literature review. J Natl Med Assoc. 2000;92(3):143-146.
- Hamid KS, Rodriguez JA, Lairmore TC. Primary splenic angiosarcoma. JSLS. 2010;14(3):431-435.
doi pubmed - Abbott RM, Levy AD, Aguilera NS, Gorospe L, Thompson WM. From the archives of the AFIP: primary vascular neoplasms of the spleen: radiologic-pathologic correlation. Radiographics. 2004;24(4):1137-1163.
doi pubmed - Abrahams AM, Hughes JH. Hemangioendothelioma of the spleen. BrJ Surg. 1952;40:68-70.
doi pubmed - Neuhauser TS, Derringer GA, Thompson LD, Fanburg-Smith JC, Miettinen M, Saaristo A, Abbondanzo SL. Splenic angiosarcoma: a clinicopathologic and immunophenotypic study of 28 cases. Mod Pathol. 2000;13(9):978-987.
doi pubmed - Myoteri D, Despoina M, Dellaportas D, Dionysios D, Ayiomamitis G, Georgios A, Strigklis K, et al. Primary angiosarcoma of the spleen: an oncological enigma. Case Rep Oncol Med. 2014;2014:193036.
doi pubmed - Hsu JT, Ueng SH, Hwang TL, Chen HM, Jan YY, Chen MF. Primary angiosarcoma of the spleen in a child with long-term survival. Pediatr Surg Int. 2007;23(8):807-810.
doi pubmed - Sordillo EM, Sordillo PP, Hajdu SI. Primary hemangiosarcoma of the spleen: report of four cases. Med Pediatr Oncol. 1981;9(4):319-324.
doi pubmed - Thompson WM, Levy AD, Aguilera NS, Gorospe L, Abbott RM. Angiosarcoma of the spleen: imaging characteristics in 12 patients. Radiology. 2005;235(1):106-115.
doi pubmed - Valbuena JR, Levenback C, Mansfield P, Liu J. Angiosarcoma of the spleen clinically presenting as metastatic ovarian cancer. A case report and review of the literature. Ann Diagn Pathol. 2005;9(5):289-292.
doi pubmed - Stutz FH, Tormey DC, Blom J. Hemangiosarcoma and pathologic rupture of the spleen. Cancer. 1973;31(5):1213-1215.
doi pubmed - Koutelidakis IM, Tsiaousis PZ, Papaziogas BT, Patsas AG, Atmatzidis SK, Atmatzidis KS. Spleen rupture due to primary angiosarcoma: a case report. J Gastrointest Cancer. 2007;38(2-4):74-77.
doi pubmed - Manouras A, Giannopoulos P, Toufektzian L, Markogiannakis H, Lagoudianakis EE, Papadima A, Papanikolaou D, et al. Splenic rupture as the presenting manifestation of primary splenic angiosarcoma in a teenage woman: a case report. J Med Case Rep. 2008;2:133.
doi pubmed - Miyata T, Fujimoto Y, Fukushima M, Torisu M, Tanaka M. Spontaneous rupture of splenic angiosarcoma: a case report of chemotherapeutic approach and review of the literature. Surg Today. 1993;23(4):370-374.
doi pubmed - Mayir B, Colak T, Dinckan A. [Spontaneous spleen rupture due to primary splenic angiosarcoma: a case report]. Ulus Travma Acil Cerrahi Derg. 2007;13(4):313-315.
- Raffel S, Hildebrandt B, Grieser C, Pahl S, Sturm I. Thrombocytopenia as first manifestation of splenic angiosarcoma. Ann Hematol. 2010;89(1):109-110.
doi pubmed - Simansky DA, Schiby G, Dreznik Z, Jacob ET. Rapid progressive dissemination of hemangiosarcoma of the spleen following spontaneous rupture. World J Surg. 1986;10(1):142-145.
doi pubmed - Maier A, Bataille F, Krenz D, Anthuber M. [Angiosarcoma as a rare differential diagnosis in spontaneous rupture of the spleen]. Chirurg. 2004;75(1):70-74.
doi pubmed - Villedieu Poignant S, Mermet L, Bousquet A, Dupont P. [A rare cause of spontaneous hemoperitoneum]. Rev Med Interne. 2000;21(9):809-811.
doi pubmed - Linan-Padilla A, Suarez-Grau JM, Valera Sanchez Z, Vazquez-Medina A, Docobo-Durantez F. [Spontaneous hemoperitoneum due to hemangiosarcoma of the spleen]. Cir Esp. 2008;84(3):171-172.
doi pubmed - Sivelli R, Piccolo D, Soliani P, Franzini C, Ziegler S, Sianesi M. [Rupture of the spleen in angiosarcoma: a case report and review of the literature]. Chir Ital. 2005;57(3):377-380.
- Winde G, Sprakel B, Bosse A, Reers B, Wendt M. Rupture of the spleen caused by primary angiosarcoma. Case report. Eur J Surg. 1991;157(3):215-217.
- Badiani R, Schaller G, Jain K, Swamy R, Gupta S. Angiosarcoma of the spleen presenting as spontaneous splenic rupture: A rare case report and review of the literature. Int J Surg Case Rep. 2013;4(9):765-767.
doi pubmed - Abdallah RA, Abdou AG, Asaad NY, Al-Sharaky DR, Alhanafy AM. Primary epithelioid angiosarcoma of spleen: a case report and review of literature. J Clin Diagn Res. 2016;10(1):ED05-07.
doi pubmed - Ozcan B, Cevener M, Kargi AO, Dikici H, Yildiz A, Ozdogan M, Gurkan A. Primary splenic angiosarcoma diagnosed after splenectomy for spontaneous rupture. Turk J Surg. 2018;34(1):68-70.
doi pubmed - Teco-Cortes JA, Navarrete-Perez JJ, Sanchez-Castro OE. Primary splenic angiosarcoma with capsular rupture and disseminated: a case report. Cir Cir. 2021;89(S2):59-63.
doi pubmed - Bilski M, Surdyka D, Pasnik I, Bilska M, Cisek P, Korona P, Szumilo J, et al. Adjuvant radiochemotherapy with a 23-month overall survival time in a patient after a surgery due to splenic hemangiosarcoma rupture: a case report with the literature review. Case Rep Oncol Med. 2018;2018:8672407.
doi pubmed - Xu B, Xie X, Zhou X, Zhai M, Yang W. Spontaneous rupture of primary splenic angiosarcoma: A case report. Oncol Lett. 2015;10(5):3271-3273.
doi pubmed - Duan YF, Jiang Y, Wu CX, Zhu F. Spontaneous rupture of primary splenic angiosarcoma: a case report and literature review. World J Surg Oncol. 2013;11:53.
doi pubmed - Wheelwright M, Spartz EJ, Skubitz K, Yousaf H, Murugan P, Harmon JV. Primary angiosarcoma of the spleen, a rare indication for splenectomy: a case report. Int J Surg Case Rep. 2021;82:105929.
doi pubmed - Mark RJ, Poen JC, Tran LM, Fu YS, Juillard GF. Angiosarcoma. A report of 67 patients and a review of the literature. Cancer. 1996;77(11):2400-2406.
doi - Naka N, Ohsawa M, Tomita Y, Kanno H, Uchida A, Myoui A, Aozasa K. Prognostic factors in angiosarcoma: a multivariate analysis of 55 cases. J Surg Oncol. 1996;61(3):170-176.
doi - Montemayor P, Caggiano V. Primary hemangiosarcoma of the spleen associated with leukocytosis and abnormal spleen scan. Int Surg. 1980;65(4):369-373.
- Hara T, Tsurumi H, Kasahara S, Ogawa K, Takada J, Imai K, Takai K, et al. Long-term survival of a patient with splenic angiosarcoma after resection, high-dose chemotherapy, and autologous peripheral blood stem cell transplantation. Intern Med. 2010;49(20):2253-2257.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.