

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website http://www.wjon.org |
Case Report
Volume 10, Number 3, June 2019, pages 153-156
Acute Myeloid Leukemia Acquiring Promyelocytic Leukemia-Retinoic Acid Receptor Alpha at Relapse
Varsha Guptaa, c, Mohammed Shariffa, Ravneet Bajwaa, Ishan Patela, Hashem A. Ayyadb, Michael J. Levitta, Peter J. Mencela, Mohammad A. Hossaina
aDepartment of Medicine, Jersey Shore University Medical Center, Hackensack Meridian Health, Neptune, NJ, USA
bDepartment of Pathology, Jersey shore University Medical Center, Neptune, NJ, USA
cCorresponding Author: Varsha Gupta, Department of Medicine, Jersey Shore University Medical Center, Hackensack Meridian School of Medicine at Seton Hall, 1945 Route 33, Neptune, NJ 07753, USA
Manuscript submitted May 31, 2019, accepted June 14, 2019
Short title: AML Acquiring PML-RARA
doi: https://doi.org/10.14740/wjon1211
| Abstract | ▴Top |
Acute promyelocytic leukemia (APL) is identified as the M3 subtype of acute myeloid leukemia (AML). APL is presently one of the most curable leukemias. We describe here a rare case of APL who presented as a relapsed disease after 1 year of chemotherapy for AML. The patient lacked t(15;17) at the initial presentation but was present later at the time of relapse. The patient attained a complete remission following treatment with all-trans retinoic acid (ATRA) and arsenic trioxide-based therapy. We discuss the possible mechanism behind secondary acquisition of promyelocytic leukemia/retinoic acid receptor alpha (PML-RARA) at relapse of AML. We also briefly discuss the clinical features, diagnosis and treatment of APL.
Keywords: Acute myeloid leukemia; All-trans retinoic acid; Acute promyelocytic leukemia
| Introduction | ▴Top |
Acute myeloid leukemia (AML) refers to a group of hematopoietic neoplasms involving cells committed to the myeloid lineage. Acute promyelocytic leukemia (APL) is identified as the M3 subtype of AML. APL was first described in late 1950s in Norway and France as a hyperacute fatal illness associated with a hemorrhagic syndrome [1]. Within three decades, complete remission with chemotherapy like daunorubicin was reported [2]. Later, Dr. Zhen Wang described the efficacy of all-trans retinoic acid (ATRA) in APL patients, in 1980s [2]. In mid 1990s, arsenic trioxide (ATO) was added to the treatment regimen [3]. Over the past few years, APL has transformed from a highly fatal disease to a highly curable one. Majority of cases are characterized by a balanced translocation between chromosome 17 and chromosome 15. This leads to an abnormal fusion protein called promyelocytic leukemia/retinoic acid receptor alpha (PML-RARA) [4].
| Case Report | ▴Top |
A 41-year-old, previously healthy, Hispanic man presented to the emergency room with complains of headache, difficulty in speech and generalized weakness. Physical exam revealed heart rate of 86/min, respiratory rate of 16/min, afebrile, and neurological exam was relevant for expressive aphasia and right-sided facial droop. Complete blood count showed pancytopenia with white blood cell (WBC) of 600/µL (4,500 - 11,000/µL), hemoglobin of 7.3 g/dL (13.2 - 17.5 g/dL) and platelet count of 14,000/µL (140,000 - 450,000/µL). Computed tomography of head showed multiple intracranial hemorrhage. Due to pancytopenia, bone marrow biopsy (BMB) (Figs. 1-4) was done which showed hypocellular marrow, and there was presence of cells resembling promyelocytes. Flow cytometry (Figs. 5 and 6) showed increased blasts (86.8% of total cells) and was positive for CD117, CD13, CD33 and CD64, negative for CD34 and HLA-Dr. Further, fluorescence in situ hybridization (FISH) study was done which was negative for t(15;17) and showed no other mutations. Karyotype was 46,XY, t(2;20)(q21;q13.1), del(14)(q24) (20). Next generation sequencing (NGS) was negative. Molecular testing using polymerase chain reaction (PCR) was not performed at that time. Diagnosis of AML was made. He was started on systemic chemotherapy with idarubicin and cytarabine. On chemotherapy day 15, bone marrow biopsy was repeated, which showed hypocellular bone marrow with no overt evidence of acute leukemia suggesting remission. However, after 1 year, he had abnormal outpatient complete blood count and so BMB was repeated, and he was found to have relapsed disease. BMB showed 31% blasts along with t(15;17) fusion. Karyotype was 46,XY, t(2;20)(q21;q13.1), del(14)(q24) (20). Diagnosis of APL was made this time. Because of the unexpected results, original AML sample was retested with FISH probe. FISH for t(15;17) was negative at initial presentation. A cryptic t(15;17) fusion was identified by FISH at relapse 1 year later, confirmed with molecular testing using PCR. The karyotype at initial presentation is identical to that at relapse which supports that this is AML acquiring t(15;17) at relapse. Patient was treated with ATRA and ATO, to which he responded very well.
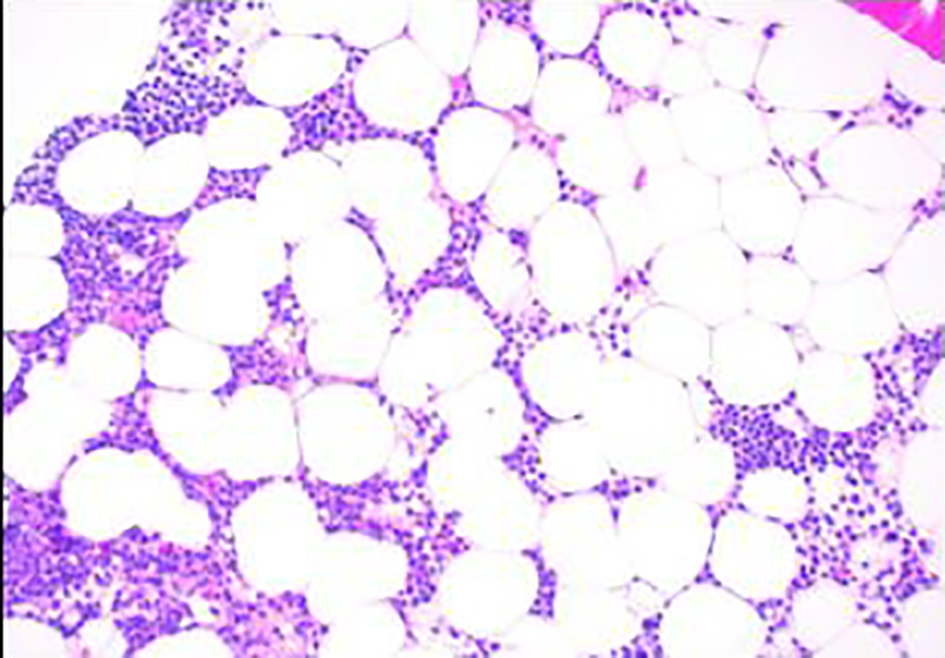 Click for large image | Figure 1. Bone marrow biopsy showing hypocellular marrow for age (× 100). |
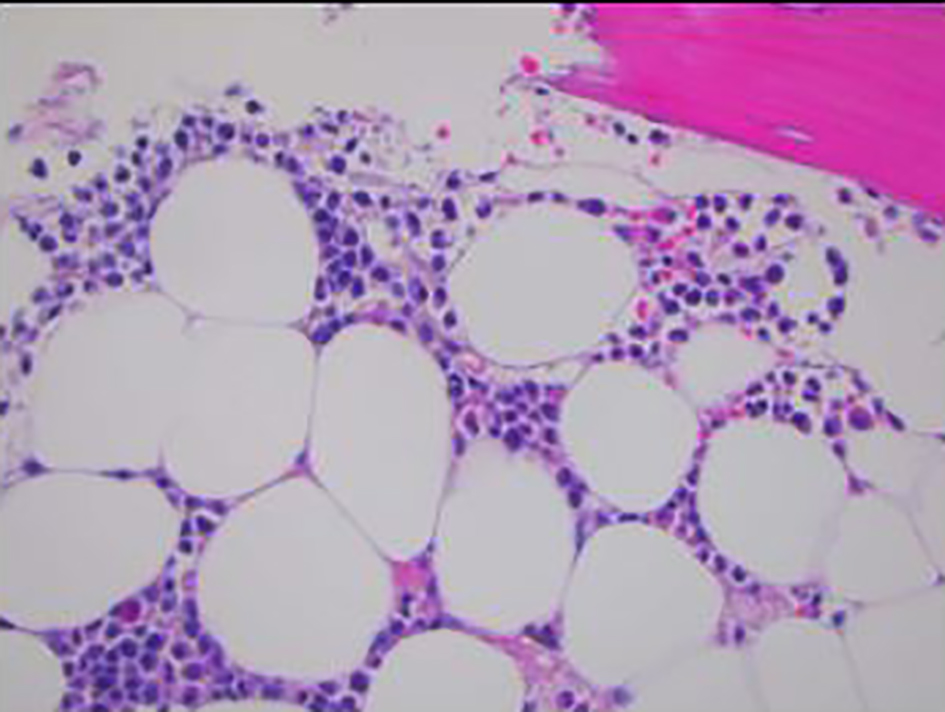 Click for large image | Figure 2. Bone marrow biopsy showing infiltration of marrow by monomorphic cells with fine chromatin and moderate amounts of granular cytoplasm (× 400). |
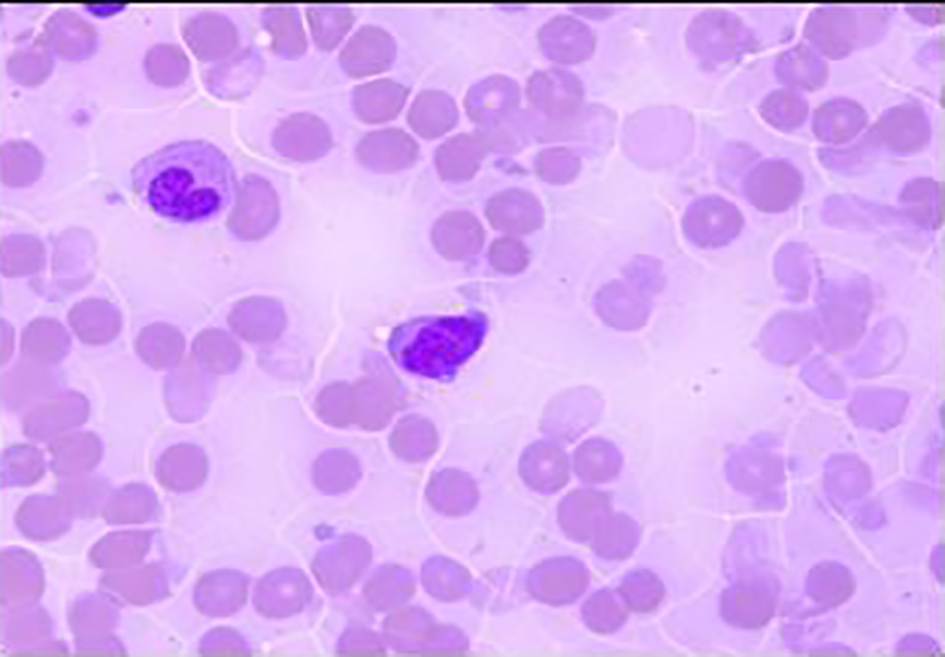 Click for large image | Figure 3. Bone marrow biopsy. Promyelocyte (blast equivalent) showing Auer rods and cytoplasmic coarse granules and fine chromatin (oil, × 1,000). |
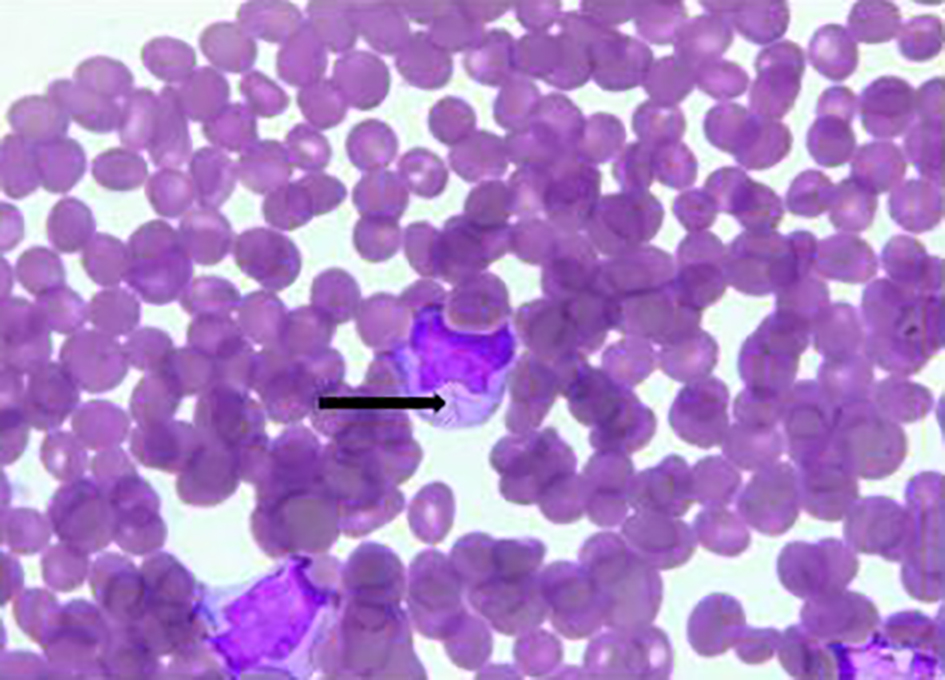 Click for large image | Figure 4. Bone marrow biopsy. Promyelocyte with Auer rod marked with black arrow. |
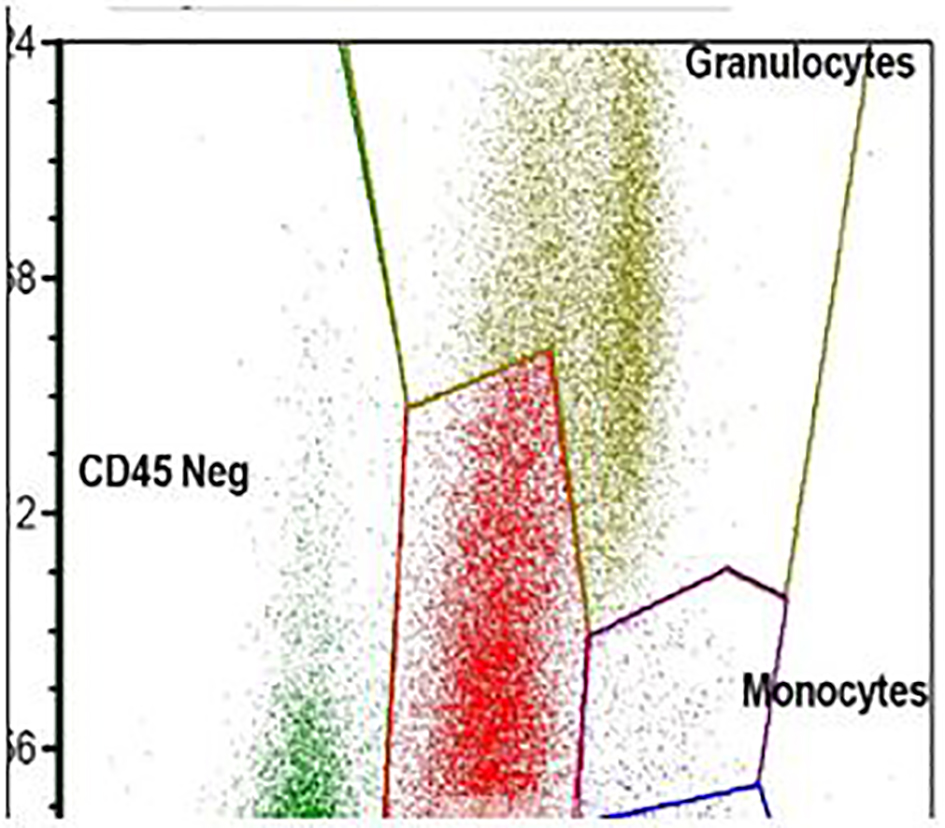 Click for large image | Figure 5. Flow cytometry. The blasts show high side scatter indicating complex cytoplasm. |
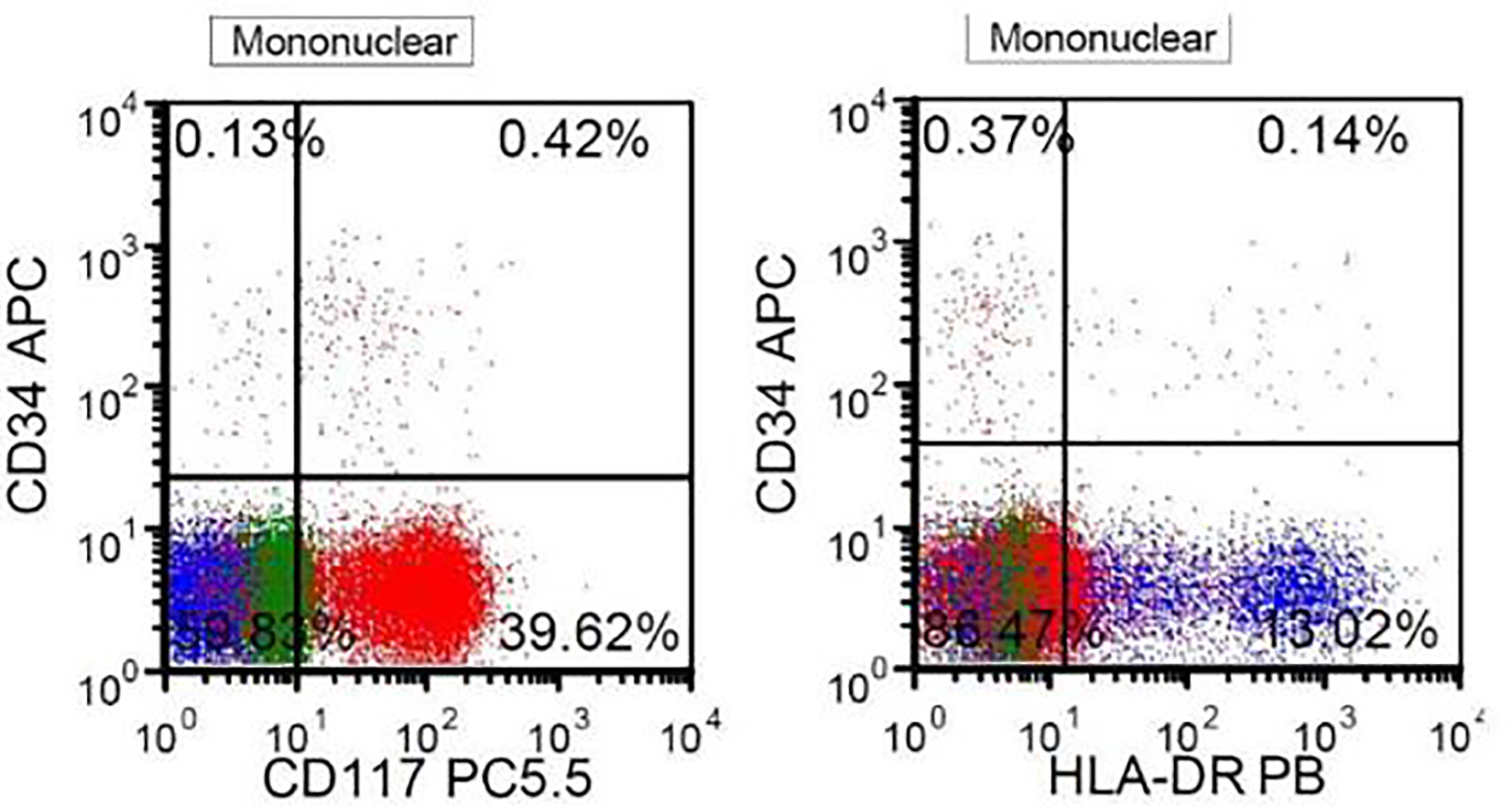 Click for large image | Figure 6. Flow cytometry. The blasts are positive for CD117 and negative for CD34 and HLA-DR, highly suggestive of APL. |
| Discussion | ▴Top |
Our patient was diagnosed to have APL at the time of relapse of AML. APL is cytogenetically characterized by the translocation in chromosomes 15 and 17 (t(15;17)). Translocation results in the fusion protein called PML-RARA which blocks the differentiation and maturation of myeloid cells at the promyelocytic stage.
Acquisition of PML-RARA as relapsed AML is rarely reported in literature. To the best of our knowledge, we found only one similar case reported by Vitale et al. In their case, it was hypothesized that there was a subclonal PML-RARA under detectable threshold and the subclone could have undergone selection during initial chemotherapy [5, 6]. Our patient also had acquisition PML-RARA as relapse of AML, after receiving 1 year of chemotherapy. Another possibility could be therapy-related APL secondary to chemotherapy [7]. APL can sometimes occur after cytotoxic therapy for another disease (e.g. breast cancer, lymphoma, other solid tumors), especially in association with the use of topoisomerase-II inhibitors such as etoposide and doxorubicin, or after radiation therapy [8]. Our patient was treated with topoisomerase-II inhibitor.
A recently published prospective analysis conducted by French-Belgian-Swiss APL group studied characteristics of de novo and secondary APLs [7]. Secondary APL arising after chemotherapy or radiotherapy constituted about 10-20% of all cases [7]. Secondary APL has also been reported more recently after mitoxantrone treatment for multiple sclerosis. As per the study, hematologic characteristics and outcomes of secondary APL were like those of de novo APL [7].
Giri et al conducted a population-based study, utilizing surveillance, epidemiology and end results (SEERs) database to compare the characteristics and survival of secondary APL and de novo APL. Study demonstrated no difference in overall survival (OS) between the two, suggesting that the underlying tumor biology of secondary APL and de novo APL is similar. At the same time, it was also suggested that both the entities can be managed similarly [9].
APL was first described in 1957 [1]. Independent risk factors for a diagnosis of APL in a patient with AML are younger age, Hispanic ethnicity and obesity [10]. Historically, as a fatal disease, now with recent advances, APL is considered a more curable disease. Patients with APL commonly present with symptoms related to complications of pancytopenia including weakness, fatigue, infection and hemorrhage. It is characterized by severe bleeding tendency due to fibrinogenemia and disseminated intravascular coagulation, accumulation of abnormal promyelocytes in bone marrow and peripheral blood [11]. Disruption of RARA function is the major cause of APL [12].
Secondary acquisition like in our patient is of a concern for a treating physician as the treatment may entirely change. Delay in administration of ATRA in cases of APL has been reported to be life-threatening. It is strongly recommended to start treatment with a differentiation agent without delay as soon as the diagnosis is suspected based on cytologic and clinical criteria, and before definitive genetic, cytogenetic, or immunostaining confirmation of the diagnosis has been made.
As addition of ATRA to chemotherapy leads to substantially improved outcomes, it is therefore very important to find better ways to diagnose cryptic oncogenes, especially related to APL. Welch et al studied the utility of whole genome sequencing (WSG) to diagnose a cryptic fusion oncogene [13]. As per their study, WSG can identify cytogenetically invisible oncogenes in a clinically relevant time frame. They described a patient with AML, who was initially being considered for allogeneic stem cell transplantation, and was instead treated with ATRA consolidation after conducting WSG [13]. Our patient underwent NGS at primary presentation which was negative. However, PCR was not performed at that time. While relapse, PCR for PML-RARA was positive.
Treatment of APL is based on risk stratification [2]. Low-risk disease is defined as WBC of ≤ 10,000/µL and high-risk disease as WBC ≥ 10,000/µL. For last few decades, ATRA + ATO-based therapy with or without chemotherapy is the backbone of management of APL. Whether or not to use chemotherapy is based on risk stratification of APL. Chemotherapy is usually omitted in low-risk disease [2]. Induction chemotherapy is still the standard of care for high-risk disease.
ATRA was introduced in 1985 [11]. It induces differentiation of leukemic promyelocytes to mature granulocytes. ATRA and ATO are synergistic in their action for inducing differentiation [2]. Using this combination therapy, complete remission (CR) rate up to 96% can be achieved. However, further studies showed that for high-risk disease inferior CR rate of 79-81% was achieved because of fatal hemorrhage and differentiation syndrome [11, 14]. Therefore, for high-risk disease, addition of cytotoxic drugs like anthracyclines is very important to prevent rapid development of leukocytosis, differentiation syndrome and relapse [15, 16].
Conclusion
It is critical to screen patients for PML-RARA fusion in all AML cases, even in the setting of relapse. APL represents a medical emergency with a high rate of early mortality. Treatment with a differentiation agent should be started without delay as soon as the diagnosis is suspected based on cytologic criteria, and even before definitive cytogenetic or molecular confirmation of the diagnosis has been made.
Acknowledgments
None to declare.
Financial Disclosure
This project was not supported by any grant or funding agencies.
Conflict of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Informed Consent
The patient described in the case report had given informed consent for the case report to be published.
Author Contributions
Each author has individually been involved and participated in drafting the manuscript and revising it critically for important intellectual content and have given final approval of the version to be published. Each has agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. MJL, PJM and MH encouraged VG, MS, RB and IP to learn about APL and secondary acquisition of PML-RARA. All authors discussed the medical literature. VG presented the idea, MS, RB and IP wrote the manuscript with input from all authors. HAA helped with pathology slides and interpretation.
| References | ▴Top |
- Bernard J. History of promyelocytic leukaemia. Leukemia. 1994;8(Suppl 2):S1-5.
- Coombs CC, Tavakkoli M, Tallman MS. Acute promyelocytic leukemia: where did we start, where are we now, and the future. Blood Cancer J. 2015;5:e304.
doi pubmed - Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89(9):3354-3360.
- De Braekeleer E, Douet-Guilbert N, De Braekeleer M. RARA fusion genes in acute promyelocytic leukemia: a review. Expert Rev Hematol. 2014;7(3):347-357.
doi pubmed - Vitale C, Jabbour E, Lu X, Yabe M, Kanagal Shamanna R, Daver N, Pemmaraju N, et al. Acute promyelocytic leukemia presented as a relapse of acute myeloid leukemia. Am J Hematol. 2016;91(4):E274-276.
doi pubmed - Stass S, Mirro J, Melvin S, Pui CH, Murphy SB, Williams D. Lineage switch in acute leukemia. Blood. 1984;64(3):701-706.
- Braun T, Cereja S, Chevret S, Raffoux E, Beaumont M, Detourmignies L, Pigneux A, et al. Evolving characteristics and outcome of secondary acute promyelocytic leukemia (APL): A prospective analysis by the French-Belgian-Swiss APL group. Cancer. 2015;121(14):2393-2399.
doi pubmed - Beaumont M, Sanz M, Carli PM, Maloisel F, Thomas X, Detourmignies L, Guerci A, et al. Therapy-related acute promyelocytic leukemia. J Clin Oncol. 2003;21(11):2123-2137.
doi pubmed - Giri S, Pathak R, Martin MG, Bhatt VR. Survival of de novo and secondary acute promyelocytic leukemia: a propensity-matched analysis of the SEER database. Leuk Lymphoma. 2016;57(2):385-391.
doi pubmed - Hillestad LK. Acute promyelocytic leukemia. Acta Med Scand. 1957;159(3):189-194.
doi pubmed - Estey E, Thall P, Kantarjian H, Pierce S, Kornblau S, Keating M. Association between increased body mass index and a diagnosis of acute promyelocytic leukemia in patients with acute myeloid leukemia. Leukemia. 1997;11(10):1661-1664.
doi pubmed - Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111(5):2505-2515.
doi pubmed - Welch JS, Westervelt P, Ding L, Larson DE, Klco JM, Kulkarni S, Wallis J, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. 2011;305(15):1577-1584.
doi pubmed - Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999;93(10):3167-3215.
- Estey E, Garcia-Manero G, Ferrajoli A, Faderl S, Verstovsek S, Jones D, Kantarjian H. Use of all-trans retinoic acid plus arsenic trioxide as an alternative to chemotherapy in untreated acute promyelocytic leukemia. Blood. 2006;107(9):3469-3473.
doi pubmed - de Botton S, Chevret S, Coiteux V, Dombret H, Sanz M, San Miguel J, Caillot D, et al. Early onset of chemotherapy can reduce the incidence of ATRA syndrome in newly diagnosed acute promyelocytic leukemia (APL) with low white blood cell counts: results from APL 93 trial. Leukemia. 2003;17(2):339-342.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.