

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Review
Volume 13, Number 6, December 2022, pages 329-336

Bimodal Age Distribution in Cancer Incidence
Shreya Desaia, Achuta K. Guddatia, b
aDivision of Hematology/Oncology, Georgia Cancer Center, Augusta University, Augusta, GA 30912, USA
bCorresponding Author: Achuta Kumar Guddati, Division of Hematology/Oncology, Georgia Cancer Center, Augusta University, Augusta, GA 30909, USA
Manuscript submitted October 5, 2021, accepted November 5, 2022, published online December 24, 2022
Short title: Bimodal Distribution in Cancer
doi: https://doi.org/10.14740/wjon1424
- Abstract
- Introduction
- Osteosarcoma
- ALL
- HL
- Craniopharyngioma
- Lung Cancer
- Kaposi Sarcoma (KS)
- Breast Cancer
- Cervical Cancer
- Conclusions
- References
| Abstract | ▴Top |
Cancer is caused by accumulation of genetic changes which include activation of protooncogenes and loss of tumor suppressor genes. The age-specific incidence of cancer in general increases with advancing age. However, some cancers exhibit a bimodal distribution. Commonly recognized cancers with bimodal age distribution include acute lymphoblastic leukemia, osteosarcoma, Hodgkin’s lymphoma, germ cell tumors and breast cancer. Delayed infection hypothesis has been used to provide explanation for the early childhood peak in leukemias and lymphomas, whereas the peak at an older age is associated with accumulation of protooncogenes and weakened immune system. Further genetic analysis and histopathological variations point to distinctly different cancers, varying genetically and histologically, which are often combined under a single category of cancers. Tumor characteristics and age distribution of these cancers varies also by population groups and has further implications on cancer screening methods. Although significant advances have been made to explain the bimodal nature of such cancers, the specific genetic mechanisms for each age distribution remain to be elucidated. Further distinction among the different cancer subtypes may lead to improvements in individual risk assessments, prevention and enhancement of treatment strategies.
Keywords: Bimodal; Hodgkin’s lymphoma; Delayed infection; Acute lymphoblastic leukemia; Craniopharyngioma; Breast cancer; Germ cell tumors; Cervical cancer
| Introduction | ▴Top |
In United States, the lifetime risk of developing cancer is significant (39.24%) [1]. Persons of all ages are susceptible to developing cancer, although the cumulative risk of cancer appears to rise with advancing age. Cancer in childhood is uncommon, representing approximately 2% of all cancer cases [1]. Subsequently, the age-specific cancer incidence rate increases with advancing age until around age 85 years [2].
Cancer is caused by accumulation of genetic changes which include the activation of protooncogenes as well as the loss of tumor suppressor genes. The exact pathogenesis by which some patients are at an increased risk of developing these genetic mutations is being investigated. Alfred Knudson’s two-hit hypothesis, however, provides insight into the genetic mutations which eventually lead to the development of cancer phenotype [3]. The two-hit hypothesis was based in the statistical analyses of children with retinoblastoma. It postulates that most tumor suppressor genes require both alleles to be inactivated. In patients with inherited retinoblastoma, the first hit is thought to be inherited via germinal cells with acquired second mutation leading to development of retinoblastoma. In children with sporadic incidence, both “hits” were thought to be acquired, causing delayed presentation [3]. The first mutation later came to be identified as the RB1 gene [4]. This two-stage model hypothesis also fits with development of other childhood and adult cancers such as Wilm’s tumor, neuroblastoma, and renal cell carcinoma [5]. However, cancer epigenetic studies have shown that cancer development is not simply explained by mutations in a single gene.
Hematopoietic tumors and central nervous system tumors comprise of nearly 65% of all childhood cancers [6]. Although these cancers are also seen in adults, childhood cancers have distinct biological and genetic differences as well as treatment outcomes. Childhood cancers are thought to have significantly fewer driver genes and have nearly 14 times lower mutation rate compared to adult tumors [7]. Approximately 5-8% of childhood cancers are thought to be due to inherited germline mutations such as TP53, APC, BRCA2, NF1 and RB1, but only 40% of the patients had family history of cancer [8]. The risk of developing childhood cancer is hypothesized by several other factors including intrauterine growth, exposure to ionizing radiation and possible role of delayed infection.
One hypothesis regarding development of childhood cancer centers on delayed infection hypothesis [9]. It is generally believed that infections acquired early in life shape the development of immune system. The delayed infection hypothesis proposes that delayed exposure to common childhood infections may cause delayed immune modulation in neonatal and infancy period, leading to abnormal function of immune system when exposed to such infectious agents later in life promoting carcinogenesis [9]. This hypothesis is supported by the study of development of acute lymphoblastic leukemia (ALL) in children. This theory may be further reinforced by the relatively lower incidence of childhood leukemia in lower income countries compared to higher income countries [10]. The risk of ALL was found to be lower among children living in poorer neighborhoods in lower income counties compared to the wealthiest neighborhoods [11]. However, research so far has failed to identify specific infections or viruses that predispose to development of ALL [9]. Additionally, studies suggest that in susceptible individuals, infections can cause abnormal immune signaling causing series of genetic alterations leading to increased malignancy susceptibility. For example, paired box 5 (PAX5) is one of the transcription factors required for correct B-cell development and is found to be altered in nearly one-third of ALL cases [12]. Mutations in PAX5 gene have been shown to lead to vulnerable population of progenitor B cells that are more prone to malignant transformation when exposed to secondary mutations (which may be precipitated by an infection) [13, 14]. The population mixing hypothesis also relies on the premise that the development of ALL may be a rare side effect of an otherwise unharmful virus in individuals with delayed exposure [15, 16]. The hypothesis suggests that immune systems of individuals in rural/isolated communities are less likely to be exposed to variety of infectious agents compared to immune systems of those living in dense population. A large scale rural-urban population mixing can thus lead to an increased level of contact between susceptible and infected individuals, leading to epidemic of certain viral infections among the rural children with delayed exposure to infections. The effect was most observed in children ages 0 - 4 years old in rural areas [17-19].
In older adults, advancing age is considered to be the most important risk factor for developing cancer [2] (Table 1 [1]). Aging is associated with many altered host functions including increased genomic instability, telomeric attrition, deregulated nutrient sensing, epigenetic alterations, mitochondrial dysfunction, cellular senescence and altered intercellular communication [20, 21]. While most mutations are corrected by the cellular mechanisms of DNA repair, the accumulated DNA damage can lead to certain mutations which cannot be repaired by the cell’s repair mechanisms. This process occurs over long periods of time, contributing not only to aging but also to rise of pro-oncogenic mutations which can then cause cancer [22]. Additionally, telomere dysfunction also appears to contribute to both cancer and aging. While short telomere length has been found to be associated with increased complications of aging such as heart disease and poor immune system, shortened telomere length can itself be associated with accelerating aging process [23]. Many cancer cells also display shortened telomere length; however, the telomerase activity is upregulated compared to normal tissues, allowing the cancer cell’s telomerase to divide beyond the Hayflick limit [24, 25]. The relationship between telomere length and cancer has been long speculated and while many studies including meta-analyses point to some evidence of shortened length as a risk factor for cancer, the results are conflicting across studies [26-28].
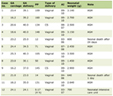 Click to view | Table 1. List of Median Age of Diagnosis of Most Common Cancers [1] |
While cancer rates in general tend to rise with advancing age, some cancers are shown to have bimodal age distribution with first peak occurring in childhood/adolescence and second peak occurring later in old age. Examples of commonly recognized cancers with bimodal age distributions include ALL, osteosarcoma, craniopharyngioma and Hodgkin’s lymphoma (HL) (Table 2 [29-39]). It is not well understood why some cancers exhibit log rising slope in incidence while others have bimodal distribution. Hypotheses for such distribution differ across different bimodal cancers.
Click to view | Table 2. Bimodal Cancers With Early and Late Median Age of Diagnosis |
| Osteosarcoma | ▴Top |
Osteosarcoma is an uncommon primary malignant tumor of the bone originating from the transformation of marrow stromal cells [29]. It has bimodal age distribution with first peak in young adults/children and second peak in older adults [29]. Childhood osteosarcoma differs from adulthood osteosarcoma in site of presentation, histology and overall survival. The osteosarcoma found in children is known as primary osteosarcoma which is commonly found in long bones near metaphyseal growth plates [40]. In adults, it is often considered a secondary neoplasm, occurring in axial sites, consisting of a sarcomatous transformation from a pre-existing bone lesion such as Paget’s disease [29]. The hypothesis behind such bimodal age distribution involves an imbalance in the supply and demand of progenitor cells [41]. The hypothesis proposes that the peak in childhood is due to the adolescent growth spurt causing imbalance in supply/demand of progenitor cells whereas the second peak among older patients is thought to be associated with increased bone turnover and remodeling process in the elderly [41]. One can then speculate whether the drop in incidence in the middle age group is due to the lack of increased progenitor cell requirement and lack of significant bone turnover.
| ALL | ▴Top |
Bimodal peaks observed in the incidence of ALL are thought to occur due to ineffective lymphoid recovery in both children and elderly. As the delayed hypothesis postulates, protections from early childhood infections can cause a dysfunctional immune response, leading to vulnerable progenitor B cells prone to malignant transformation. One assumes that this risk decreases with age, leading to drop in the incidence of ALL after childhood [30]. However, incidence of ALL again peaks in the elderly, and is then thought to be due to failing immune suppressor mechanisms [12]. The bimodal distribution of the cancer may also be due to different genomic changes causing similar phenotype of cancer across age groups. For example, adult patients with ALL have reduced prevalence of ETV-RUNX1 compared to childhood ALL which although produces the same phenotype of ALL, can have significant impact on prognosis and treatment outcomes [42].
| HL | ▴Top |
The delayed infection hypothesis is also used to explain the bimodal distribution in HL. Classic HL is a heterogenous disease with histological, genetic and virological differences. The initial peak is seen among adults 21 - 30 years and second peak in patients aged > 70 years, although the patterns of incidence vary among levels of socioeconomic development, gender and race [31, 43, 44]. There are distinct histological differences in HL cases among young adults and older patients with younger adults more likely to have Epstein-Barr virus (EBV)-negative, nodular sclerosing subtype whereas older adults are noted to have higher occurrence of EBV-positive, mixed cellularity subtype [31, 45, 46]. Additionally, lymphocyte depletion subtype is mostly observed in older adults [45]. In young adults, the increased incidence of HL is hypothesized to be due to delayed exposure to common, non-EBV childhood infections [43]. Several studies have shown the relative risk of HL decreased with increasing sibship size and late birth order which likely contributes to earlier exposure to common infections via siblings [44, 47, 48]. Thus, HL may be a rare abnormal immunological response due to delayed exposure to common illnesses in childhood causing delayed maturation of cellular immunity. The second peak among older adults is thought to be due to loss of immunological control of latent EBV infection [49]. This is supported by the higher EBV positivity in older HL patients [31, 45, 46, 49]. HL in this population may be a rare effect of common EBV infection with increasing probability of oncogenesis with advancing age. Additionally, genetic differences are noted among older patients compared to young adults. Older age is found to be associated with decreased FOXP3 regulatory T cells and increased granzyme-B+ cells (P = 0.02 and P = 0.01, respectively) with poor prognosis in older patients [50].
| Craniopharyngioma | ▴Top |
Indeed, bimodal peaks in other cancers may also be explained by the heterogeneity of tumors with different biological and embryonic differences. Another example is that of craniopharyngioma which has a bimodal age distribution with peak incidence rates in children of ages 5 - 14 years and adults ages 50 - 75 years [32]. Craniopharyngiomas are rare tumors of the craniopharyngeal duct with two distinct histological subtypes: adamantinomatous craniopharyngioma (ACP) and papillary craniopharyngioma (PCP) [51]. It is primarily ACP which carries the bimodal age distribution as PCP is mainly found in adults in their 50 - 60 years of age [51]. The embryonic theory states that ACP arises from malignant transformation of the remnant ectodermal cells which failed to involute after formation of the craniopharyngeal duct in embryogenesis [52]. Whereas the metaplastic theory argues that craniopharyngioma develops from metaplasia of adenohypophyseal cells leading to the formation of squamous cell nests. This theory is supported by the increasing presence of metaplastic nests with advanced age [51, 52]. What remains unclear is why the middle age group is not predisposed to metaplasia or malignant transformation of remnant ectodermal cells. One hypothesis may pertain to an increased presence of functional tumor suppressor genes in young adults compared to the pediatric or elderly population.
| Lung Cancer | ▴Top |
While not exhibiting bimodal age distribution in childhood and old age, lung cancer serves as a good example in which multiple histologically different cancers may be classified under one disease umbrella. Different mutations are found in lung cancer of young patients versus older patients [53]. A rare disease called the midline carcinoma of children and young adults with NUT rearrangement was first described in the early 1990s and identified as an extension of thymic carcinoma or laryngeal cancer [54]. Further analysis of this disease revealed a translocation t(15;19)(q13, p13.1) which was associated with a highly lethal course in young people despite a healthy body [55]. The translocation results in either a BRD4-NUT fusion protein or a pairing with an unknown gene leading to the NUT variant [54]. This entity is now recognized as an aggressive squamous cell epithelial cancer arising from the midline of the body. Despite its recognition as a single entity, now called NUT midline carcinoma, due to its low prevalence, it remains largely unknown. Additionally, many of such cancers may possibly continue to be misidentified into other cancer categories. However, misclassification into any of the other categories due to a lack of complete evaluation from the unavailability of resources or lack of awareness about this disease can cause skewing of the data for other cancer groups. NUT midline carcinomas have been identified to occur primarily in the adolescent and early adult age group, and misclassification of these cancers may falsely cause a new earlier mode peak when the incidence is charted [54].
| Kaposi Sarcoma (KS) | ▴Top |
In United States, the incidence of KS also follows a bimodal distribution with peaks noted among ages 30 - 36 years and in patients > 70 years [33]. It is generally understood that classic KS tumors emerge as a result of compromised immune system in patients previously exposed to human gamma herpesvirus 8 (HHV8) [56]. Among young patients, this immunocompromised situation is thought to occur from prevalence of human immunodeficiency virus (HIV) infection and development of acquired immune deficiency syndrome (AIDS). The proposed mechanisms by which KS risk is increased include increased HHV8 viral replication and reactivation of latent HHV8 infection [56]. Among older patients, the incidence of KS appears to be unassociated with AIDS and includes classic variant initially described among patients of Mediterranean descent as well as iatrogenic variants [57]. Mediterranean patients have been found to have high HHV8 prevalence, with incidence of KS supported by the hypothesis that it develops in the setting of age-related decline in immune function [58]. Additionally, iatrogenic causes of KS may also contribute to higher incidence of KS in older population, likely due to a longer duration of immunosuppressive medication in combination with natural decline in immune function [57]. It can thus be postulated that middle age patients are less likely to develop KS in spite of HHV8 prevalence due to lower prevalence of AIDS and non-age-related immune dysfunction.
| Breast Cancer | ▴Top |
Breast cancer also shows bimodal age distribution at diagnosis with early incidence peak at 45 years and late peak at 65 years [34]. This distribution is present across estrogen receptor (ER) status, tumor characteristics and histological subtypes with significant proportional variations, making breast cancer extremely heterogenous disease [59]. The exception to the bimodal age distribution is medullary carcinoma of the breast, associated with germline mutations in BRCA1, which follows a single distribution [60]. Studies have shown basal-like, human epidermal growth factor receptor 2 (HER2)- enriched and luminal B cancers to have predominantly early-onset peaks whereas luminal A cancers had late-onset peak and minor early onset peak [59]. Additionally, higher proportions of estrogen receptor (ER)- protein and ESR1-RNA categories were seen among the patients with late age of onset. Such molecular characterizations have led to hypothesis of two distinct causal pathways of carcinogenesis from distinct progenitor cell types [60]. This includes derivation of tumors from stem cells committed to luminal differentiation with ER expression versus the development of tumors from stem cells committed to basal differentiation lacking ER expression [34, 60].
Several hormonal factors impact an individual’s risk of developing breast cancer, including the age of first menarche, age of menopause as well as age of first pregnancy [61]. While the rates of breast cancer are lower in younger women, the tumors characteristics were found to be more likely to be aggressive with higher incidence of distant metastases compared to the older patients [61]. An analysis of the SEER database showed a higher percentage of black women with breast cancer at a younger age compared to an older age (15.32% vs. 10.26%). They were also noted to be less likely to have hormone positive cancers [61]. This aligns with similar tumor characteristics seen in Jamaican women [62]. A retrospective analysis showed that younger patients made a greater cohort of breast cancer diagnoses in Jamaica compared to United States, with the lower mean age of diagnosis being 54 years [62]. Additionally, luminal A subtype was less prevalent in Jamaica compared to overall rate seen on the US Databases (49% vs. 73%) [62]. This variation in age distribution of breast cancer is again observed in Nigeria where younger premenopausal women again accounted for the majority of breast cancer diagnoses 63]. Up to 47.4% of the patients had triple negative subtype which is associated with increased risk of tumor invasion and poor prognosis [63]. This is in contrast to tumor subtypes of patients in United States, where the rates of aggressive subtypes are considerably lower [63].
Given the diverse age distribution of cancer and tumor heterogeneity among different populations, screening programs can have significant implications. The current US Preventive Services Task Force recommendations for breast cancer recommend routine screening in women over the age of 50 with the decision to screen women between ages 40 and 49 years old to be left between the patient and the physician [64]. It is unclear whether the same screening strategies are beneficial in patients of all backgrounds. Asian women in Japan, for example, have the highest incidence of breast cancer in the 45 - 49 years age group with lower incidence in the older age group [65]. Several differences in the distribution of risk factors including lower prevalence of obesity and lower use of hormone replacement therapies may explain the lower rate of postmenopausal breast cancer among Asian women [65]. Additionally, breast tissues of Japanese women were reported to have less ER-positivity, possibly explaining the lower prevalence of postmenopausal ER- positive breast cancer in Japanese women compared to Western populations [65]. It remains to be seen whether Asian populations within western countries exhibit the same tumor characteristics and age distribution as the women residing in Asian countries. Further stratification and understanding of breast cancer tumor biology is warranted to individualize risk assessment, prevention and treatment strategies in high-risk patients.
| Cervical Cancer | ▴Top |
Cervical cancer also shows bimodal age distribution with the first peak at ages 30 - 34 years and second peak at 65 - 69 years of age [35]. Interestingly, this age distribution varies 5 - 15 years apart from the bimodal distribution of age-specific human papillomavirus (HPV) infection prevalence at 25 years and 45 years of age [66]. The time lapse is likely explained by the time to malignant transformation after infection by HPV. Such bimodal distribution of HPV infection is seen globally which is considered atypical as HPV prevalence is expected to decline with age [66-68]. Multiple hypotheses may explain the bimodal occurrence of HPV prevalence and cervical cancer. The first peak of HPV infection in younger cohort is possibly due to primary exposure to HPV after sexual initiation, likelihood of multiple sexual partners and possible lack of adaptive immune responses [35]. Higher rates of HPV infection in older women may be due to viral persistence or reactivation of latent HPV due to physiological and immunological dysregulation at the menopause transition [35, 69]. Additionally, lack of screening in the elderly age group may also contribute towards a higher incidence of cervical cancer in the elderly women, leading to a second peak. While one may expect HPV infection rates and subsequently cervical cancer rates to reduce as more women are vaccinated, some studies have shown that older women are more likely to be infected with nonvalent HPV strains compared to younger women, which may again explain the rise in incidence [70]. Effective screening among all age groups remains key for improving outcomes across all age groups.
| Conclusions | ▴Top |
Although significant advances have been made to explain the bimodal nature of certain cancers, questions remain regarding the clinical application of the hypotheses. Bimodal cancers represent a unique opportunity for understanding carcinogenesis, impact of the immune system and the effect of environmental factors which cause the development of widely heterogeneous group of cancers that are often classified under a single category or are misclassified. Further stratifications of these cancers and enhanced understanding of tumor characteristics on a sub-population level may lead to improvements in individual risk assessments, prevention and enhancement of treatment strategies.
Acknowledgments
None to declare.
Financial Disclosure
The authors declare that there was no funding for this study.
Conflict of Interest
None to declare.
Author Contributions
Conception and design: AG. Analysis and interpretation: SD and AG. Drafting and formatting: SD and AG. Critical revision and final editing: SD and AG.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, Ruhl J (eds). SEER Cancer Statistics Review, 1975-2018. 2021: Bethesda, MD: National Cancer Institute. Available from: https://seer.cancer.gov/csr/1975_2018/.
- Thakkar JP, McCarthy BJ, Villano JL. Age-specific cancer incidence rates increase through the oldest age groups. Am J Med Sci. 2014;348(1):65-70.
doi pubmed - Knudson AG, Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820-823.
doi pubmed - Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, Dryja TP. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature. 1986;323(6089):643-646.
doi pubmed - Maher ER, Yates JR, Ferguson-Smith MA. Statistical analysis of the two stage mutation model in von Hippel-Lindau disease, and in sporadic cerebellar haemangioblastoma and renal cell carcinoma. J Med Genet. 1990;27(5):311-314.
doi pubmed - Murphy MF, Bithell JF, Stiller CA, Kendall GM, O'Neill KA. Childhood and adult cancers: contrasts and commonalities. Maturitas. 2013;76(1):95-98.
doi pubmed - Savary C, Kim A, Lespagnol A, Gandemer V, Pellier I, Andrieu C, Pages G, et al. Depicting the genetic architecture of pediatric cancers through an integrative gene network approach. Sci Rep. 2020;10(1):1224.
doi pubmed - Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373(24):2336-2346.
doi pubmed - Greaves M. Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer. 2006;6(3):193-203.
doi pubmed - Howard SC, Metzger ML, Wilimas JA, Quintana Y, Pui CH, Robison LL, Ribeiro RC. Childhood cancer epidemiology in low-income countries. Cancer. 2008;112(3):461-472.
doi pubmed - Ribeiro KB, Buffler PA, Metayer C. Socioeconomic status and childhood acute lymphocytic leukemia incidence in Sao Paulo, Brazil. Int J Cancer. 2008;123(8):1907-1912.
doi pubmed - Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, Hagiwara K, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51(2):296-307.
doi pubmed - Auer F, Ruschendorf F, Gombert M, Husemann P, Ginzel S, Izraeli S, Harit M, et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia. 2014;28(5):1136-1138.
doi pubmed - Martin-Lorenzo A, Hauer J, Vicente-Duenas C, Auer F, Gonzalez-Herrero I, Garcia-Ramirez I, Ginzel S, et al. Infection Exposure is a Causal Factor in B-cell Precursor Acute Lymphoblastic Leukemia as a Result of Pax5-Inherited Susceptibility. Cancer Discov. 2015;5(12):1328-1343.
doi pubmed - Kinlen LJ. Epidemiological evidence for an infective basis in childhood leukaemia. Br J Cancer. 1995;71(1):1-5.
doi pubmed - Kinlen L. Evidence for an infective cause of childhood leukaemia: comparison of a Scottish new town with nuclear reprocessing sites in Britain. Lancet. 1988;2(8624):1323-1327.
doi pubmed - Kinlen L, Doll R. Population mixing and childhood leukaemia: Fallon and other US clusters. Br J Cancer. 2004;91(1):1-3.
doi pubmed - Kinlen LJ, Bramald S. Paternal occupational contact level and childhood leukaemia in rural Scotland: a case-control study. Br J Cancer. 2001;84(7):1002-1007.
doi pubmed - Koushik A, King WD, McLaughlin JR. An ecologic study of childhood leukemia and population mixing in Ontario, Canada. Cancer Causes Control. 2001;12(6):483-490.
doi pubmed - Burkhalter MD, Rudolph KL, Sperka T. Genome instability of ageing stem cells—Induction and defence mechanisms. Ageing Res Rev. 2015;23(Pt A):29-36.
doi pubmed - Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194-1217.
doi pubmed - Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Budovsky A, Yanai H, Fraifeld VE. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res Rev. 2013;12(2):661-684.
doi pubmed - Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96(5):701-712.
doi pubmed - Blackburn EH, Epel ES, Lin J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science. 2015;350(6265):1193-1198.
doi pubmed - Shay JW, Wright WE. Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol. 2000;1(1):72-76.
doi pubmed - Ma H, Zhou Z, Wei S, Liu Z, Pooley KA, Dunning AM, Svenson U, et al. Shortened telomere length is associated with increased risk of cancer: a meta-analysis. PLoS One. 2011;6(6):e20466.
doi pubmed - Wentzensen IM, Mirabello L, Pfeiffer RM, Savage SA. The association of telomere length and cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2011;20(6):1238-1250.
doi pubmed - Telomeres Mendelian Randomization Collaboration, Haycock PC, Burgess S, Nounu A, Zheng J, Okoli GN, Bowden J, et al. Association between telomere length and risk of cancer and non-neoplastic diseases: a mendelian randomization study. JAMA Oncol. 2017;3(5):636-651.
- Ottaviani G, Jaffe N. The epidemiology of osteosarcoma. Cancer Treat Res. 2009;152:3-13.
doi pubmed - Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood. 2012;119(1):34-43.
doi pubmed - Shenoy P, Maggioncalda A, Malik N, Flowers CR. Incidence patterns and outcomes for hodgkin lymphoma patients in the United States. Adv Hematol. 2011;2011:725219.
doi pubmed - Bunin GR, Surawicz TS, Witman PA, Preston-Martin S, Davis F, Bruner JM. The descriptive epidemiology of craniopharyngioma. J Neurosurg. 1998;89(4):547-551.
doi pubmed - Dittmer DP, Damania B. Kaposi's Sarcoma-Associated Herpesvirus (KSHV)-associated disease in the AIDS patient: An Update. Cancer Treat Res. 2019;177:63-80.
doi pubmed - Anderson WF, Pfeiffer RM, Dores GM, Sherman ME. Comparison of age distribution patterns for different histopathologic types of breast carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15(10):1899-1905.
doi pubmed - Sun LL, Jin Q, Li H, Zhou XR, Song ZQ, Cheng XM, Tao T, et al. Population-based study on the prevalence of and risk factors for human papillomavirus infection in Qujing of Yunnan province, Southwest China. Virol J. 2012;9:153.
doi pubmed - Bray F, Haugen M, Moger TA, Tretli S, Aalen OO, Grotmol T. Age-incidence curves of nasopharyngeal carcinoma worldwide: bimodality in low-risk populations and aetiologic implications. Cancer Epidemiol Biomarkers Prev. 2008;17(9):2356-2365.
doi pubmed - Dumont B, Lemelle L, Cordero C, Couloigner V, Bernard S, Cardoen L, Brisse HJ, et al. Esthesioneuroblastoma in children, adolescents and young adults. Bull Cancer. 2020;107(9):934-945.
doi pubmed - Ramai D, Ofosu A, Lai JK, Gao ZH, Adler DG. Fibrolamellar Hepatocellular Carcinoma: A Population-Based Observational Study. Dig Dis Sci. 2021;66(1):308-314.
doi pubmed - Fonseca A, Frazier AL, Shaikh F. Germ cell tumors in adolescents and young adults. J Oncol Pract. 2019;15(8):433-441.
doi pubmed - Wagner ER, Luther G, Zhu G, Luo Q, Shi Q, Kim SH, Gao JL, et al. Defective osteogenic differentiation in the development of osteosarcoma. Sarcoma. 2011;2011:325238.
doi pubmed - Richardson RB. Age-specific bone tumour incidence rates are governed by stem cell exhaustion influencing the supply and demand of progenitor cells. Mech Ageing Dev. 2014;139:31-40.
doi pubmed - Roberts KG. Genetics and prognosis of ALL in children vs adults. Hematology Am Soc Hematol Educ Program. 2018;2018(1):137-145.
doi pubmed - Grotmol T, Bray F, Holte H, Haugen M, Kunz L, Tretli S, Aalen OO, et al. Frailty modeling of the bimodal age-incidence of Hodgkin lymphoma in the Nordic countries. Cancer Epidemiol Biomarkers Prev. 2011;20(7):1350-1357.
doi pubmed - Hjalgrim H, Soegaard SH, Hjalgrim LL, Rostgaard K. Childhood use of antimicrobials and risk of Hodgkin lymphoma: a Danish register-based cohort study. Blood Adv. 2019;3(9):1489-1492.
doi pubmed - Engert A, Ballova V, Haverkamp H, Pfistner B, Josting A, Duhmke E, Muller-Hermelink K, et al. Hodgkin's lymphoma in elderly patients: a comprehensive retrospective analysis from the German Hodgkin's Study Group. J Clin Oncol. 2005;23(22):5052-5060.
doi pubmed - Jarrett RF, Stark GL, White J, Angus B, Alexander FE, Krajewski AS, Freeland J, et al. Impact of tumor Epstein-Barr virus status on presenting features and outcome in age-defined subgroups of patients with classic Hodgkin lymphoma: a population-based study. Blood. 2005;106(7):2444-2451.
doi pubmed - Westergaard T, Melbye M, Pedersen JB, Frisch M, Olsen JH, Andersen PK. Birth order, sibship size and risk of Hodgkin's disease in children and young adults: a population-based study of 31 million person-years. Int J Cancer. 1997;72(6):977-981.
doi - Gutensohn N, Cole P. Childhood social environment and Hodgkin's disease. N Engl J Med. 1981;304(3):135-140.
doi pubmed - Maggioncalda A, Malik N, Shenoy P, Smith M, Sinha R, Flowers CR. Clinical, molecular, and environmental risk factors for hodgkin lymphoma. Adv Hematol. 2011;2011:736261.
doi pubmed - Kelley TW, Pohlman B, Elson P, Hsi ED. The ratio of FOXP3+ regulatory T cells to granzyme B+ cytotoxic T/NK cells predicts prognosis in classical Hodgkin lymphoma and is independent of bcl-2 and MAL expression. Am J Clin Pathol. 2007;128(6):958-965.
doi pubmed - Muller HL, Merchant TE, Warmuth-Metz M, Martinez-Barbera JP, Puget S. Craniopharyngioma. Nat Rev Dis Primers. 2019;5(1):75.
doi pubmed - Yachnis AT. Chapter 4 - craniopharyngioma: embryology, pathology, and molecular aspects. In: Kenning TJ, Evans JJBTC, Editors. Academic Press: Boston. 2015. p. 95-105.
doi - Kreuzer M, Kreienbrock L, Muller KM, Gerken M, Wichmann E. Histologic types of lung carcinoma and age at onset. Cancer. 1999;85(9):1958-1965.
doi - French CA, Kutok JL, Faquin WC, Toretsky JA, Antonescu CR, Griffin CA, Nose V, et al. Midline carcinoma of children and young adults with NUT rearrangement. J Clin Oncol. 2004;22(20):4135-4139.
doi pubmed - Vargas SO, French CA, Faul PN, Fletcher JA, Davis IJ, Dal Cin P, Perez-Atayde AR. Upper respiratory tract carcinoma with chromosomal translocation 15;19: evidence for a distinct disease entity of young patients with a rapidly fatal course. Cancer. 2001;92(5):1195-1203.
doi pubmed - Ganem D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest. 2010;120(4):939-949.
doi pubmed - Shiels MS, Pfeiffer RM, Hall HI, Li J, Goedert JJ, Morton LM, Hartge P, et al. Proportions of Kaposi sarcoma, selected non-Hodgkin lymphomas, and cervical cancer in the United States occurring in persons with AIDS, 1980-2007. JAMA. 2011;305(14):1450-1459.
doi pubmed - Iscovich J, Boffetta P, Franceschi S, Azizi E, Sarid R. Classic kaposi sarcoma: epidemiology and risk factors. Cancer. 2000;88(3):500-517.
doi - Allott EH, Shan Y, Chen M, Sun X, Garcia-Recio S, Kirk EL, Olshan AF, et al. Bimodal age distribution at diagnosis in breast cancer persists across molecular and genomic classifications. Breast Cancer Res Treat. 2020;179(1):185-195.
doi pubmed - Anderson WF, et al. Bimodal breast cancer incidence patterns provide support for a dualistic model of mammary carcinogenesis. Journal of Clinical Oncology. 2006;24(18_suppl):595-595.
doi - Nasim Z, Girtain C, Gupta V, Patel I, Hossain MA. Breast cancer incidence and behavior in younger patients: a study from the surveillance, epidemiology and end results database. World J Oncol. 2020;11(3):88-97.
doi pubmed - Copeland J, Oyedeji A, Powell N, Cherian CJ, Tokumaru Y, Murthy V, Takabe K, et al. Breast cancer in jamaica: stage, grade and molecular subtype distributions across age blocks, the implications for screening and treatment. World J Oncol. 2021;12(4):93-103.
doi pubmed - Adeniji AA, Dawodu OO, Habeebu MY, Oyekan AO, Bashir MA, Martin MG, Keshinro SO, et al. Distribution of breast cancer subtypes among nigerian women and correlation to the risk factors and clinicopathological characteristics. World J Oncol. 2020;11(4):165-172.
doi pubmed - Siu AL, Force USPST. Screening for Breast Cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med. 2016;164(4):279-296.
doi pubmed - Tsuchida J, Nagahashi M, Rashid OM, Takabe K, Wakai T. At what age should screening mammography be recommended for Asian women? Cancer Med. 2015;4(7):1136-1144.
doi pubmed - Alsbeih GA, Al-Harbi NM, Bin Judia SS, Khoja HA, Shoukri MM, Tulbah AM. Reduced rate of human papillomavirus infection and genetic overtransmission of TP53 72C polymorphic variant lower cervical cancer incidence. Cancer. 2017;123(13):2459-2466.
doi pubmed - Mutombo AB, Benoy I, Tozin R, Bogers J, Van Geertruyden JP, Jacquemyn Y. Prevalence and distribution of human papillomavirus genotypes among women in Kinshasa, the Democratic Republic of the Congo. J Glob Oncol. 2019;5:1-9.
doi pubmed - Kim MA, Oh JK, Chay DB, Park DC, Kim SM, Kang ES, Kim JH, et al. Prevalence and seroprevalence of high-risk human papillomavirus infection. Obstet Gynecol. 2010;116(4):932-940.
doi pubmed - Mandelblatt J. Squamous cell cancer of the cervix, immune senescence and HPV: is cervical cancer an age-related neoplasm? Adv Exp Med Biol. 1993;330:13-26.
doi pubmed - Hammer A, Mejlgaard E, Gravitt P, Hogdall E, Christiansen P, Steiniche T, Blaakaer J. HPV genotype distribution in older Danish women undergoing surgery due to cervical cancer. Acta Obstet Gynecol Scand. 2015;94(11):1262-1268.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.