

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Review
Volume 15, Number 1, February 2024, pages 1-13

Autotaxin and Lysophosphatidate Signaling: Prime Targets for Mitigating Therapy Resistance in Breast Cancer
Matthew G.K. Benescha, h, Xiaoyun Tangb, David N. Brindleyb ![]() , Kazuaki Takabea, c, d, e, f, g, h
, Kazuaki Takabea, c, d, e, f, g, h ![]()
aDepartment of Surgical Oncology, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14263, USA
bCancer Research Institute of Northern Alberta, Department of Biochemistry, University of Alberta, Edmonton, AB T6G 2H7, Canada
cDepartment of Breast Surgery and Oncology, Tokyo Medical University, Tokyo 160-8402, Japan
dDepartment of Gastroenterological Surgery, Yokohama City University Graduate School of Medicine, Yokohama 236-0004, Japan
eDivision of Digestive and General Surgery, Niigata University Graduate School of Medical and Dental Sciences, Niigata 951-8520, Japan
fDepartment of Breast Surgery, Fukushima Medical University School of Medicine, Fukushima 960-1295, Japan
gDepartment of Surgery, University at Buffalo Jacobs School of Medicine and Biomedical Sciences, State University of New York, Buffalo, NY 14263, USA
hCorresponding Author: Matthew Benesch, Department of Surgical Oncology, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14263, USA; Kazuaki Takabe, Department of Surgical Oncology, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14263, USA
Manuscript submitted November 5, 2023, accepted December 29, 2023, published online January 20, 2024
Short title: Autotaxin, Lysophosphatidate, and Breast Cancer
doi: https://doi.org/10.14740/wjon1762
- Abstract
- Introduction: Overview of Autotaxin and Lysophosphatidate Signaling in Breast Cancer
- Insights Into ATX Production in the Breast Tumor Microenvironment
- Overview of Recent Insights Into LPAR and LPP Function in Breast Cancer
- Summary of the Current State of LPA Pathway Pharmacological Interventions
- Future Perspective on Modern ATX and LPA Signaling Translational Research
- References
| Abstract | ▴Top |
Overcoming and preventing cancer therapy resistance is the most pressing challenge in modern breast cancer management. Consequently, most modern breast cancer research is aimed at understanding and blocking these therapy resistance mechanisms. One increasingly promising therapeutic target is the autotaxin (ATX)-lysophosphatidate (LPA)-lipid phosphate phosphatase (LPP) axis. Extracellular LPA, produced from albumin-bound lysophosphatidylcholine by ATX and degraded by the ecto-activity of the LPPs, is a potent cell-signaling mediator of tumor growth, invasion, angiogenesis, immune evasion, and resistance to cancer treatment modalities. LPA signaling in the post-natal organism has central roles in physiological wound healing, but these mechanisms are subverted to fuel pathogenesis in diseases that arise from chronic inflammatory processes, including cancer. Over the last 10 years, our understanding of the role of LPA signaling in the breast tumor microenvironment has begun to mature. Tumor-promoting inflammation in breast cancer leads to increased ATX production within the tumor microenvironment. This results in increased local concentrations of LPA that are maintained in part by decreased overall cancer cell LPP expression that would otherwise more rapidly break it down. LPA signaling through six G-protein-coupled LPA receptors expressed by cancer cells can then activate virtually every known tumorigenic pathway. Consequently, to target therapy resistance and tumor growth mediated by LPA signaling, multiple inhibitors against the LPA signaling axis are entering clinical trials. In this review, we summarize recent developments in LPA breast cancer biology, and illustrate how these novel therapeutics against the LPA signaling pathway may be excellent adjuncts to extend the efficacy of evolving breast cancer treatments.
Keywords: Adipose tissue; Adjuvant therapy; Chemoresistance; Cytokines; ENPP2; Metastasis; Lysophosphatidic acid; Tumor models
| Introduction: Overview of Autotaxin and Lysophosphatidate Signaling in Breast Cancer | ▴Top |
As the most common cancer in women with a 1 in 8 lifetime risk, breast cancer persists as a perplexing disease to manage, particularly in the context of either relapsed or metastatic disease [1, 2]. The primary challenge to improving patient survival is overcoming treatment resistance that develops via mechanisms either intrinsic or acquired to advanced cancers [3]. The underpinnings of most current breast cancer research are typically directed at targeting these pathways with novel adjunct therapies to either desensitize or stave off resistance development or increase the therapeutic index of treatment regimens [3, 4].
One such mediator of therapy resistance is a potent extracellular signaling molecule called lysophosphatidate (LPA), which is primarily produced from serum lysophosphatidylcholine (LPC) by the phospholipase D activity of a secreted enzyme called autotaxin (ATX) (gene name ENPP2) [5]. ATX is believed to interact with cell surface integrins and other extracellular binding molecules to concentrate LPA in the local environment [6]. LPA mediates a plethora of physiological processes primarily involved in embryogenesis, tissue repair, and tissue regeneration by signaling through six known G-protein coupled receptors (LPAR1-6). Additionally, LPA signaling contributes to multiple hallmarks of cancer [5, 7]. Extracellular LPA is broken down by the ecto-activities of three membrane bound lipid phosphate phosphatases (LPPs) (gene names PLPP1-3) to monoacylglycerols (MAG), which typically lack signaling properties except for 2-arachidonoylglycerol [8].
ATX was first identified in 1992 in the cell media of cultured melanoma cells as an “autocrine motility factor” [9]. Ten years later, it was discovered that ATX exerted its biological function via its hydrolysis of LPC to LPA [10, 11]. Around this time and thereafter, a general paradigm for LPA signaling in cancer began to unfold centered on increased tumor proliferation, cancer cell migration, angiogenesis, and cancer treatment survival and resistance [12]. Primarily through cell culture investigations, cancer cells were shown to potentiate LPA signaling by either increasing both ATX secretion and LPAR receptor expression or decreasing ecto-LPP activity primarily through downregulation of LPP1 and LPP3 [8, 13, 14]. The overall net effect would result in an increased pool of extracellular LPA that can activate or augment virtually every known pathway implicated in tumorigenesis [5, 15]. Further, ATX is one of the 40 - 50 most upregulated genes in both locally invasive and metastatic tumors [16, 17]. As such, the ATX-LPA-LPP signaling pathway became an enticing area for therapeutic research not only in cancer, but also in multiple disease processes mediated by chronic inflammation [18]. This is because LPA signaling, while physiologically underpinning acute wound healing inflammatory processes, can readily be perturbed into a state of persistent signaling in these pathological conditions where “wounds” do not heal [5, 18].
ATX is overexpressed in many cancers compared to normal or benign tissue from the same organs, such as in melanoma, glioblastoma multiforme, hepatocellular carcinoma, and thyroid carcinoma [19]. However, most breast cancer cells produce little or virtually no ATX compared to normal breast tissue, but these cells still display increased tumorgenicity in response to LPA signaling. As in most cancers, downregulation of ecto-LPP expression increases LPA signaling [20-22]. Seminal work supporting the relevance of LPA signaling in breast cancer in biological systems was demonstrated in mammary mouse tumor virus (MMTV) mice with transgenic overexpression of ATX or LPAR1, LPAR2, or LPAR3 [23]. These mice develop breast tumorigenesis and subsequent metastasis in 32-53% of mice compared to no tumor development in wild type mice over the study period [23]. However, bridging the disconnect between the negligible ATX production findings in breast cancer cell culture and these animal results coincided with the concept of the tumor microenvironment coming into vogue in cancer research [24, 25].
We showed in an orthotopic mouse 4T1/Balb/c model, which is syngeneic and immunocompetent, that the growing breast tumor induced ATX expression in the surrounding mammary fat pad through the effects of LPA in activating NF-κB and upregulation of inflammatory cytokine production [21, 22]. These results led to the concept of there being a feedforward ATX-LPA-inflammatory cycle that drives tumor progression. Additionally, treatment with a potent oral ATX inhibitor not only slowed initial tumor growth and subsequent lung metastasis, but also decreased the concentrations of multiple inflammatory cytokines in the tumor [21, 22]. The 4T1/Balb/c breast cancer model is highly inflammatory and metastatic compared to the use of E0771 breast cancer cells in syngeneic C57BL/6 mice where metastasis is not observed [26]. However, inhibiting ATX with the oral ATX inhibitor IOA-289 decreased tumor growth in this model and this was accompanied by decreases in the concentrations of the inflammatory cytokines chemokine CXC ligand (CXCL)10, CC chemokine ligand 2 (CCL2), and CXCL9 in the plasma and leukemia inhibitory factor (LIF), transforming growth factor (TGF)β1, TGFβ2, and prolactin in the tumors [26].
We further validated this paracrine-induced inflammatory model of ATX production between breast tumor cells and the tumor stroma in human breast tumor specimens by demonstrating an immunohistochemical gradient of approximately two-fold increased ATX and cytokine staining in tumor adjacent stroma compared to patient-matched breast stroma distant from the tumor [21].
Preliminary clinical studies additionally have suggested a linkage between stromal ATX expression and breast tumor aggressiveness. Among the first of these investigations, demonstrated by immunohistochemistry, it was found that stromal ATX was upregulated in 50% of advanced stage breast tumors compared to only 17.6% of stage II cancers and in no stage I specimens examined [27]. Serum studies comparing ATX levels in 112 breast cancer patients to 50 healthy participants showed significantly increased concentrations in cancer patients, with stepwise increases with progressive clinical disease [28]. We showed similar results in the plasma of mice with advanced breast cancer [29]. Mechanistically, we also demonstrated that LPA could exert feedback product inhibition on further ATX transcription, thereby regulating its own extracellular secretion [29]. However, this inhibition can be overcome by increased ATX expression and secretion in response to increased inflammatory cytokine production. This occurs in the context of acute inflammation with physiological wound healing, or chronic inflammation in pathological conditions like cancer, often described as wounds that do not heal (Fig. 1) [4, 5, 8, 29-34]. This finding, along with our model of tumor microenvironment ATX production in breast cancer, has since been replicated by other investigators [35, 36].
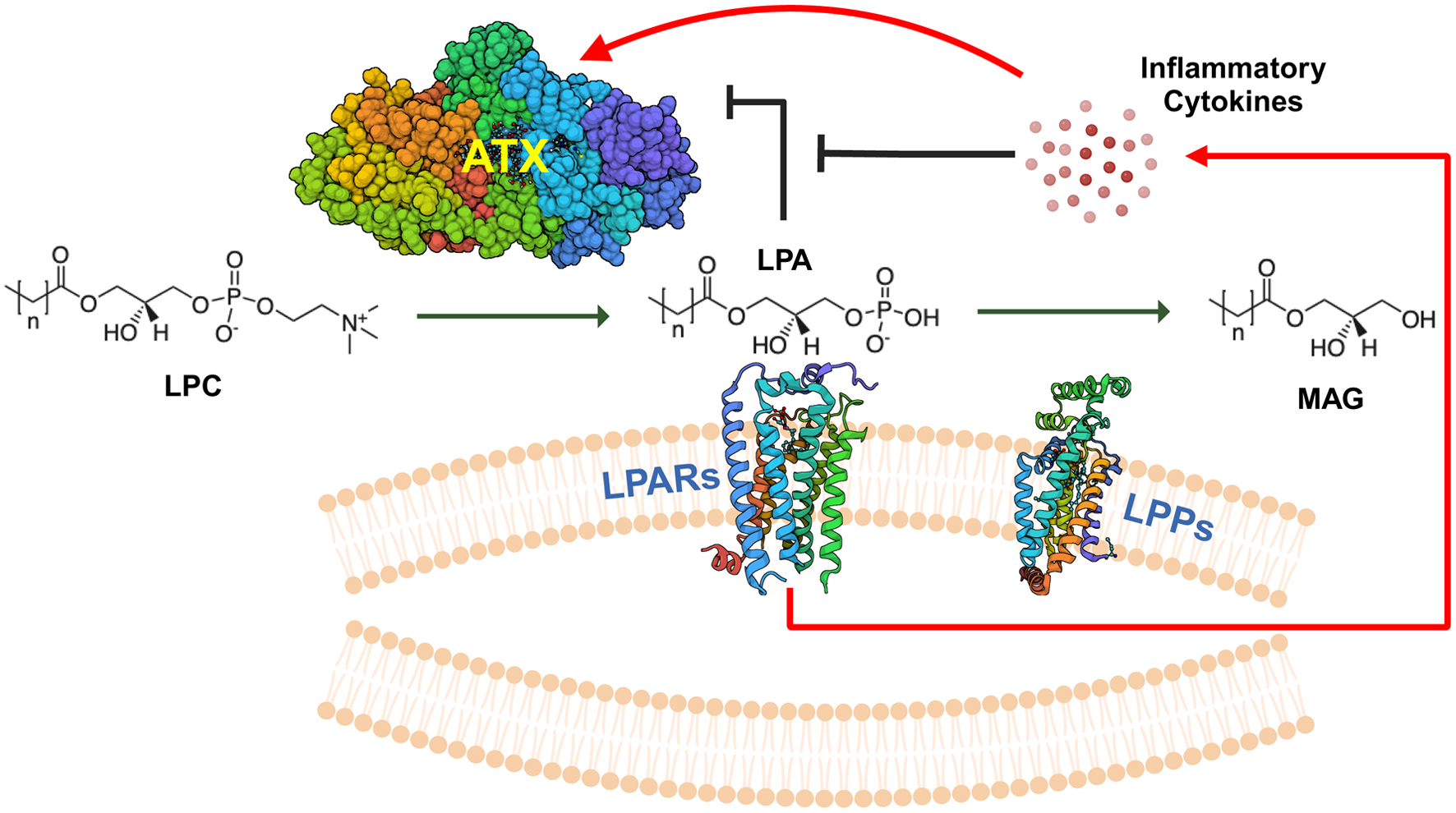 Click for large image | Figure 1. Overview of ATX-LPAR-LPP signaling. Autotaxin (ATX) is a secreted 125-kDa glycoprotein with lysophospholipase D activity, which generates lysophosphatidate (LPA) from lysophosphatidylcholine (LPC), the most abundant phospholipid in the plasma at concentrations of about 200 µM in human beings [32]. Plasma LPA levels are typically in the 100 - 300 nM range [33]. LPA signals through at least six G-protein coupled receptors (LPARs) to elicit intracellular effects. Signaling through these receptors may be either redundant or antagonistic, depending on the coupling between the receptor and the heterotrimeric G-protein [5, 34]. LPA is degraded by the ecto-activity of three lipid phosphate phosphatases (LPPs) [8]. Cancers can increase the tumorigenic effects of LPA signaling by increasing local concentrations of extracellular LPA through either increasing ATX secretion (either by the cancer cells themselves or induction in the tumor stroma) or by decreasing ecto-LPP activity levels, and by increasing LPAR levels [4]. LPA can exert feedback inhibition on ATX transcription to decrease further LPA production [29]. However, signaling mediated by inflammatory cytokines can increase ATX protein expression in the tumor microenvironment, which can overcome this feedback inhibition [29]. MAG: monoacylglycerols. |
In this review, we summarize a large body of growing evidence on new insights into the ATX-LPA-LPP axis in breast cancer tumor biology, particularly as they relate to novel discoveries related to the tumor microenvironment. We additionally provide expert commentary into ongoing translational and clinical research in breast cancer that is setting the stage for more clinical trials with inhibitors of LPA signaling. Ultimately, blocking the cross-talk of LPA signaling between breast cancer cells and their surrounding supportive microenvironment is predicted to adjunctly extend the efficacy of cancer therapy, by both limiting tumor growth and metastasis, and mitigating activation of survival pathways involved in therapy resistance.
| Insights Into ATX Production in the Breast Tumor Microenvironment | ▴Top |
Although multiple groups have demonstrated that ATX in the breast tumor microenvironment is undoubtably induced in the tumor stroma [22, 35, 36], our understanding of the cross talk and the players involved in this communication is evolving. We know from murine conditional knockout studies that about 40% of plasma ATX is produced via adipocytes [37, 38]. Adipose tissue composes from 4% to 38% of the total breast by weight [39]. These findings would favor breast adipose tissue as a possible primary source of ATX in breast tumor development. Our early work in murine tumor models showed that breast cancer cells produce virtually no ATX but instead induce ATX expression in adjacent tumor stroma [21, 22, 29]. Based on this, Schmid et al isolated and cultured epithelial and mesenchymal tumor cells from four luminal B or triple negative mammary carcinomas from human patients, as well as adipose-derived stem cells from healthy breast tissue, tumor adjacent tissue, or tumor distant tissue (at least 10 cm from the tumor) [36]. At both the mRNA and protein level, adipose-derived stem cells from any source had higher ATX levels than mesenchymal tumor cells, and epithelial cells expressed virtually no ATX [36]. After adipogenic differentiation, ATX mRNA and protein levels were nearly two-fold higher in cultures from tumor-adjacent adipose-derived stem cells compared to either cultures form tumor distant tissue or healthy tissue [36]. While this study supports adipose tissue as a source of ATX in breast tumors, these results are not derived from a model of the intact tumor microenvironment.
To study the cells that produce ATX for breast tumor growth, our group crossed immunocompetent C57BL/6 mice carrying adipocyte-specific ATX knockout with MMTV-PyMT mice [26]. As predicted [37], the adipocyte-specific ATX knockout displayed an approximately 37% reduction in total plasma ATX concentration [26]. However, the timing of spontaneous palpable breast tumors, time for tumors to reach 1 cm, and number of lung micrometastases were the same between control and knockout mice groups [26]. Similarly, C57BL/6 mice were injected with syngeneic E0771 breast cancer cells into mammary fat pads. Again, the ATX knockout mice had the same tumor growth rate as the controls, but tumor growth was slowed with the oral ATX inhibitor, IOA-289 [26]. E0771 tumors were then digested and sorted by flow cytometry into E0771 cells, CD45+ leukocytes, fibroblasts, and other cells. Relative to tumor-derived E0771 cells, leukocytes and fibroblasts expressed about 16-fold more, and other cells about five-fold more, ATX mRNA [26]. Therefore, from these investigations, these breast tumors are dependent on tumor microenvironment-derived ATX for tumor growth, however, this ATX does not necessarily have to be sourced from breast adipocytes.
To answer the question where ATX is produced in the human breast tumor microenvironment, we analyzed expression patterns in over 5,000 non-metastatic breast cancers from the TCGA, METABRIC, and GSE96058 (SCAN-B) databases [40]. Consistent with animal model studies [21, 22], tumor ATX mRNA levels were lower than in normal breast tissue (Fig. 2a) [15, 40-42]. Tumors from all three databases were cybersorted with the xCell algorithm, and high ATX expression correlated most strongly with adipocyte, fibroblast, and endothelial cell fractions [40]. We then verified these results with single cell RNA sequencing results, which demonstrated the highest expression in endothelial cells, followed by myeloid cells and cancer-associated fibroblasts (Fig. 2b) [15, 26, 40-42]. Whether there is a temporal relevance to tumor pathogenesis as to which cell populations express ATX remains an open question.
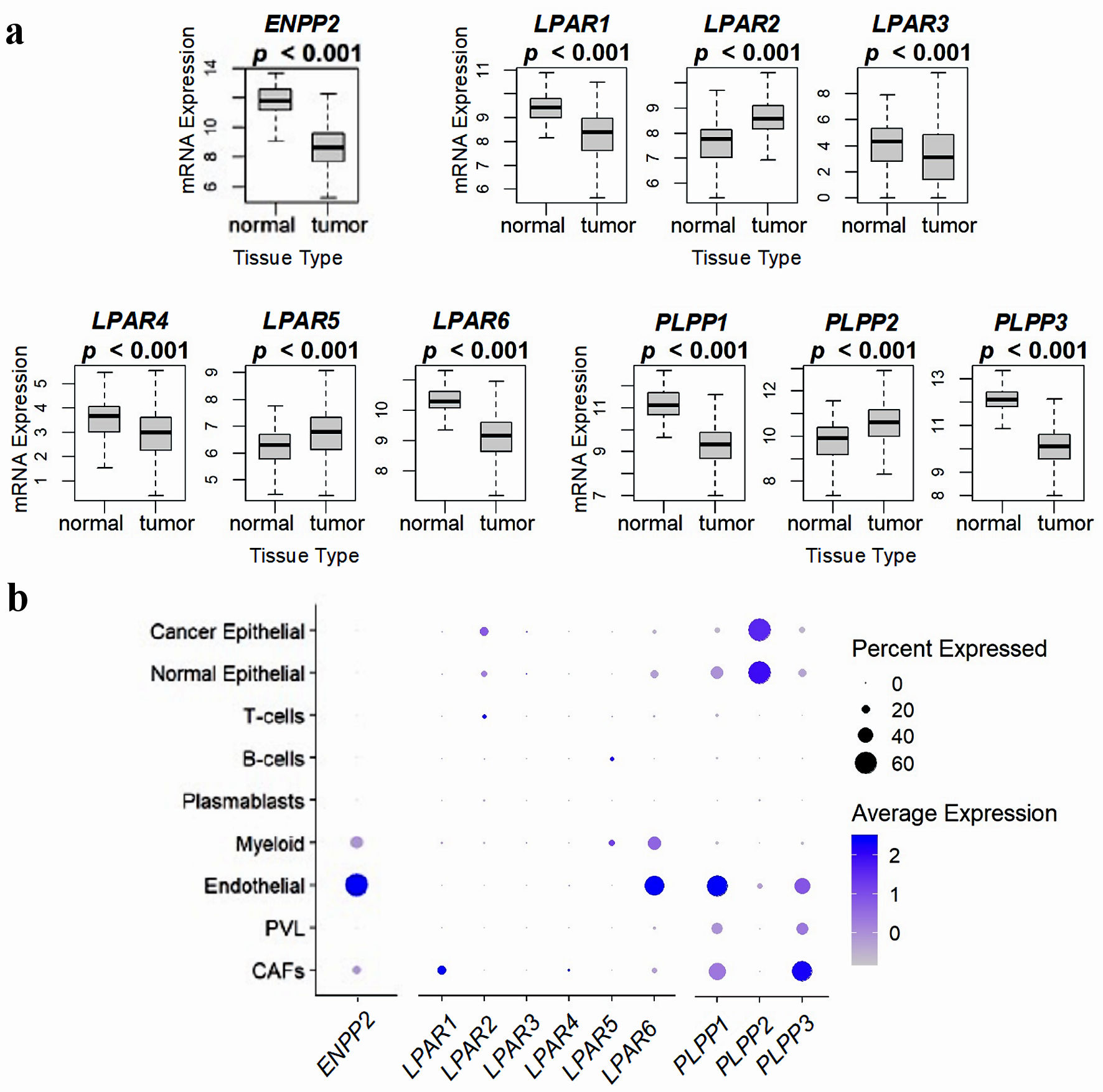 Click for large image | Figure 2. Comparison of ATX, LPARs, and LPPs expression in normal breast tissue and whole breast tumors, and via breast tumor single cell RNA sequencing. (a) mRNA expression from 114 normal breast tissues and 1,090 whole breast cancer tumors from the TCGA database. Results are plotted as box plots, with the bolded center bar representing the median, the lower and upper bounds the 25th and 75th percentiles, respectively, and the lower and upper tails the minimum and maximum values, respectively. (b) Single-cell RNA sequencing results from the cohort described in [42], comprised of 26 breast tumors (11 ER+ HER2-, five HER2+, and 10 TNBC), for a total of 130,246 single cells, to demonstrate which cell populations in the tumor express these genes. Percent expressed (circle size) refers to the percent of the total gene expression for the whole tumor by cell type, and color refers to the average gene expression. Figures were reproduced from [15, 40, 41] with permission. ATX: autotaxin; LPP: lipid phosphate phosphatase; ER: estrogen receptor; HER: human epidermal growth factor receptor; TNBC: triple-negative breast cancer. |
In the context of cancer overall, ATX is typically considered a pro-tumorigenic enzyme. This perspective is likely too simplistic, because ATX/LPA signaling influences a myriad of biological processes. To illustrate this, ATX knockout in mice is embryonically lethal at day 9.5 secondary to neural tube and vascular defects [43, 44]. After birth, ATX orchestrates wound healing via platelet aggregation and the growth and migration of fibroblasts, endothelial cells, keratinocytes, and leukocytes [5]. Hence, if ATX is a wound healing enzyme, it is possible that in very early breast cancer, ATX could have anti-tumor properties, but as the tumor grows and learns to evade the immune system, it highjacks cross-talk in the tumor microenvironment to subvert ATX/LPA signaling for progressive tumorigenesis. When we examined this intriguing notion in early breast tumors from the TCGA, METABRIC, and GSE96058 (SCAN-B) databases, high ATX-expressing tumors (when compared to low ATX-expressing tumors dichotomized at the median expression level) correlated significantly with decreased tumor mutational burden, lower Ki67 scores, increased immune cell population infiltration, increased immune cytolytic scores, and overall survival trends with hazard ratios between 0.75 and 0.80 [40]. While our study did not have enough locally advanced/metastatic tumors to draw definitive conclusions for a pro-tumor switch with more aggressive disease, gene set enrichment analysis demonstrated enriched gene signatures in high ATX expressing tumors for pro-tumor survival gene sets [40]. These sets included signatures for angiogenesis, hypoxia, reactive oxygen species, xenobiotic metabolism, inflammatory-mediated signaling, and stemness gene sets including the epithelial mesenchymal transition [40]. Hence, breast tumors high in ATX expression appear to be primed to mediate tumorigenesis in a maladaptive manner by using the physiological wound healing mechanisms and subverting them to promote tumor growth and metastasis.
ENPP2 (the ATX gene) is amplified in multiple cancer types, particularly melanoma, and the degree of amplification correlates positively with worse progression-free survival [45]. In a large survey of over 2,000 breast tumors, ENPP2 amplification was found in 26.6% of ductal carcinomas and 14.5% of lobular carcinomas [4, 46]. Despite this, relative to normal breast tissues, the ENPP2 promoter in breast cancer cells was highly methylated, even in stage 1 breast cancers [47]. This methylation pattern hence appeared to occur early in breast cancer development and did not significantly change with more advanced disease [47]. If fact, analysis of data from TCGA demonstrate that ENPP2 is of one of the 66 most hypermethylated genes in stage 1-3 breast cancer [48]. Whether this methylation is a protective event of the host organism to suppress tumor development is unknown, but these studies demonstrate that despite breast cancer cells displaying the propensity to overexpress ATX, they are prevented from doing so even at early stages, and instead must rely on inducing ATX in the surrounding tumor stroma to induce LPA-mediated tumorigenesis [49, 50].
| Overview of Recent Insights Into LPAR and LPP Function in Breast Cancer | ▴Top |
Biological systems tend to make use of multiple receptors to the same activating ligand to fine tune signaling depending on the temporal and spatial needs of the microenvironment [51]. Consequently, it is not surprising that LPA signaling through any of the six LPARs can have complementary, synergic, or antagonistic effects depending on the G-protein complex to which the receptors interact [5, 15, 52]. LPAR1-3 are ubiquitously expressed across most tissue types and belong to the Edg (endothelial differentiation gene) family [24]. LPAR4-6 have not been studied as extensively and belong to the P2Y puringenic receptor family [53]. Unlike ATX, all murine individual LPAR knockouts are viable [52, 54], as are all double LPAR1-3 knockouts and the triple LPAR1-3 knockout [32, 55]. Of the known double knockouts, only the LPAR4/LPAR6 knockout is embryonically lethal secondary to angiogenesis malformations [56].
Similar to our recent analysis of ATX expression in the breast tumor microenvironment, we performed the same analysis on over 5,000 breast tumors from the TCGA, METABRIC, and GSE96058 (SCAN-B) databases, with additional single cell RNA-sequencing validation. Compared to normal breast tissue, breast tumors have significantly lower LPAR1,3,4, and 6 mRNA levels, whereas the opposite is true for LPAR2 and LPAR5 (Fig. 2a) [15, 40-42]. Uniquely, LPAR2 expression was highest in cancer epithelial cells, and high tumor LPAR2 expression correlated strongly with increased tumor grade and mutational burden, triple-negative breast cancer (TNBC) hormone status, and decreased survival [15]. These findings have been validated in breast cancer cell culture, where LPAR2 inhibition alone was capable of limiting TNBC growth via blockage of autocrine production of proinflammatory cytokines interleukin (IL)-6, IL-8, and CXCL1 in an NF-κB-dependent manner [57]. Additionally, when ATX, LPAR1, LPAR2, and LPAR3 were overexpressed in an MMTV model, tumorigenicity was highest in the LPAR2 mice at 52.8%, followed by ATX at 50.0%, LPAR3 at 42.3%, and LPAR1 at 32.0% [23].
Infiltration and activation of T cells, particularly CD8+ cytotoxic T cells, is a critical requirement for immune-mediated tumor eradication [58]. In recent years, LPAR5-mediated signaling has emerged as a unique suppressor of immune cell infiltration into the tumor microenvironment by suppressing the CD8+ T cell function via inhibition of intracellular Ca2+ mobilization and ERK activation [59], inhibition of antigen-specific CD8+ T cell proliferation following activation [60], and impediment of CD8+ T cell function by impeding granzyme B granule exocytosis [59, 61]. Though LPAR5 levels are increased in bulk breast tumor compared to normal tissue (Fig. 2a) [15, 40-42], LPAR5 gene expression did not significantly affect survival parameters [15]. However, selective inhibition of LPAR5 might have synergistic effects with immunotherapy approaches in improving cytolytic responses, particularly in TNBC, for which immunotherapy adjuncts are now being used [62]. An alternative approach is to use ATX inhibitors such as IOA-289, which increases CD8+ T cell infiltration in breast tumors in mouse models [26, 63].
Consistent with our findings (Fig. 2a) [15, 40-42], LPAR6 is known to be downregulated particularly in human epidermal growth factor receptor (HER)2+ and TNBC, and decreased expression in all subtypes correlated to decreased survival [64-66]. LPAR6 has been proposed to function as a tumor suppressor in part through the formation of E2F family complexes capable of inducing cell cycle arrest [64]. Mechanistically, the microRNA miR-27a-3p acts as an upstream positive regulator of LPAR6 transcription and is also suppressed in breast tumors compared to normal or benign breast tissues [64]. These findings might provide a mechanistic explanation for our observation that ATX expression in early breast tumors correlated to better survival [40]. It is possible that LPA signaling in this situation through LPAR6 contributes to cell cycle arrest, and with disease progression, LPAR6 expression is repressed, resulting in loss of tumor repression via this pathway. It is possible that LPA signaling through LPAR4 could have a similar phenotype (Fig. 2a) [15, 40-42], as one study in colon cancer cell cultures showed that cell motile activities were markedly stimulated by either LPAR4 or LPAR6 knockdown [67]. This phenomenon has not been reported in breast cancers, though we did show that the cell cycling gene sets enriched in low-expressing LPAR6 tumors are nearly identical to those seen in low-expressing LPAR4 tumors [15].
It is well known that breast tumors (and other cancers, particularly ovarian) express low tumor cell LPP1 and LPP3 and high LPP2 concentrations relative to normal breast tissue [8, 68]. Breast and ovarian murine allograft and xenograft models with implanted cancer cells overexpressing either LPP1 or LPP3 results in tumors with slower growth and decreased subsequent metastasis [69, 70]. Low LPP1 expression in breast cancer cells is known to increase cyclin D1 and D3 levels and concentrations of matrix metalloproteinases, culminating in increased rates of cell division [71]. While LPP1 and LPP3 function primarily via ecto-cell activity to regulate LPA concentrations, LPP2 probably functions primarily endocellularly [8, 12]. LPP2 overexpression in many cancer types increases the rate of S-phase entry though c-Myc transcription factor upregulation [72-74], and LPP2 knockout in MDA-MB-231 breast cancer cells decreases tumor growth and lung metastasis in mouse xenografts [74]. We confirmed these phenotypic findings in three large cohorts of human breast tumors (Fig. 2a) [15, 40-42], and through single cell RNA-seq analysis, we demonstrated that most tumor LPP1 and LPP3 was expressed in the tumor stroma, while LPP2 was primarily found in the cancer cells [41]. Besides c-Myc pathways, E2F pathways were additionally upregulated in high LPP2-expressing tumors on gene set enrichment analysis and correlated with worse tumor grade and decreased overall survival [41]. Combined, these results suggest that the development of LPP2-specific inhibitors to block endo-LPP catalytic targets may have therapeutic benefits.
| Summary of the Current State of LPA Pathway Pharmacological Interventions | ▴Top |
As the initiator of the LPA signaling axis, most therapeutic development against LPA signaling has been towards designing potent and orally bioavailable ATX inhibitors. The first oral ATX inhibitor tested in murine breast cancer models was ONO-8430506. We showed that this inhibitor slowed initial tumor growth and lung metastasis in an orthotopic and immunocompetent murine breast cancer model (4T1/Balb/c), largely through reducing LPA-mediated cytokine expression in the tumor microenvironment [21, 22, 29]. We and others additionally showed that ATX inhibition with ONO-8430506 improved the effectiveness of doxorubicin [75] and paclitaxel [76]. Our group later used the ATX inhibitor GLPG1690, subsequently known as ziritaxestat, to show increased efficiency of doxorubicin in mouse breast cancer models and decrease the percentage of Ki-67 positive cells and increase rates of apoptosis with concurrent radiotherapy [77]. Ziritaxestat was the first ATX to enter clinical trials, eventually culminating in two identically designed phase III double-blind and placebo-controlled trials that combined ziritaxestat with standard of care treatments for idiopathic pulmonary fibrosis (IPF) (ISABELA 1, ziritaxestat 600 mg daily; ISABELA 2, ziritaxestat 200 mg daily) [78, 79]. The trials were eventually terminated early due to lack of efficacy over standard of care and potential safety concerns in the high dose group [78]. Other non-competitive tunnel-binding inhibitors like BLD-0409 (cudetaxestat), and BBT-877 are currently in phase II trials for IPF, with estimated completion dates of mid-2024 [80-82].
ATX substrate binding-pocket targeting inhibitors are currently divided into five different classes [83]. Class I are lipid-like or orthosteric inhibitors, with the early inhibitor PF-8380 being the prototypical inhibitor of this class [84, 85]. None of these compounds have entered advanced clinical trials, secondary to their off-target effects related to their high partition coefficient [86]. More modern inhibitors are non-carboxylic and non-lipid in design [86]. Class II inhibitors target the hydrophobic pocket (including GRI-918013, none of which are in trials), obstructing binding of LPC to ATX [83]. Class III inhibitors are allosteric tunnel inhibitors, which act non-competitively to deter the release and transport of hydrolyzed LPA, which includes the inhibitor PAT-347 by PharaAkea Inc. [87]. Class IV inhibitors, described as pocket-tunnel hybrids, include ziritaxestat, and essentially represent inhibitors with combined features of both class II and III inhibitors [83]. Finally, steroid-derived hybrids which function as both orthosteric and allosteric inhibitors that do not form interactions with the catalytic sites have recently been described and represent the latest class (class V) to be proposed [88]. In general, the most potent ATX inhibitors belong to classes II, III, IV, given how closely they interact or block access to the zinc ion-containing active site [86]. The history of ATX inhibition design has been summarized in several good reviews [83, 89-93].
Within the last year, two new phase 1 clinic trials have been initiated for first in-class inhibitors. IOA-289, a mixed type II/IV inhibitor, is a potent mitigator of lung fibrosis biomarkers, and inhibits tumor outgrowth and lung and bone metastasis in both 4T1/Balb/c and E0771/C57BL/6 murine breast cancer models [26, 63]. Another recent study has shown this inhibitor to reduce cell growth and migration of gastrointestinal tract cell lines in both two-dimensional (2D) and three-dimensional (3D) in vitro models [94]. This inhibitor is currently being investigated in a phase 1b, open label, dose-escalation study either alone or in combination with gemcitabine/nab-paclitaxel in patients with metastatic pancreatic cancer, the first ATX inhibitor to enter a clinical trial for cancer [95]. Finally, a phase 1 study in healthy volunteers examined the pharmacology and tolerability of a new inhibitor FTP-198, which demonstrated superior inhibition of LPA18:2 formation compared to ziritaxestat, without safety or tolerability concerns [96]. Another practical approach to decreasing the activation of the ATX-LPA-inflammatory cycle is to decrease tumor necrosis factor (TNF)-α-induced inflammation with infliximab, a monoclonal antibody that binds to TNF-α to attenuate its inflammatory actions and activation of ATX expression. This approach was effective in decreasing lung metastasis by 60% in the 4T1/Balb/c breast cancer model [97].
To date, no LPAR inhibitors have been tested in cancer patients, though numerous LPAR1 inhibitors, including BMS-986020 and BMS-986278, have reached phase 2 trial status for IPF [98, 99], and SAR100842 for systemic sclerosis [100]; but to date, their efficacy has not been well established. Most of these inhibitors were trialed in the late 2010s, and interest, particularly in the IPF field, has shifted to ATX inhibitors with ziritaxestat and cudetaxestat. Despite this, multiple agonists and antagonists for LPAR1-5, and at least two agonists against LPAR6, have been developed for experimental use, and are well reviewed elsewhere [66, 101, 102].
Currently, there are no known LPP2 inhibitors, which could have potential therapeutic benefit in breast cancer, as previously described [41, 103]. We have previously reported that tetracycline can increase LPP1 and LPP3 protein stability in breast cancer cells [104]. Additionally, LPP1 expression is inducible by dexamethasone treatment via inflammatory signaling repression, and this is accompanied by a decrease in the expressions of ATX and LPAR1 [105]. Although doxycycline and dexamethasone are exerting these effects on the LPPs though nonspecific functions and they may have limited therapeutic utility, the results do provide proof of principle that LPP1 and LPP3 levels can be pharmacologically manipulated to decrease the impact of chronic activation of the ATX-LPA-inflammatory cycle [103]. Pharmacological induction of LPP1/LPP3 expression to increase the turnover of LPA in the breast tumor microenvironment also remains an intriguing area for further research.
| Future Perspective on Modern ATX and LPA Signaling Translational Research | ▴Top |
LPA production by extracellular ATX represents the initiating event of a massive signaling cascade that interacts at some level with every major pathway in tumor biology. As to be expected for such a potent mediator of complex cancer signal transduction, our simplistic model of ATX autocrine overproduction by cancer cells, which then signals though LPARs to elicit pro-tumorigenesis phenotypes, has evolved, particularly in the context of breast cancer. Over the last 10 years, our understanding of LPA signaling in breast cancer has become deeply interwoven into the elaborate and nuanced additional complexities that describe the myriad of cellular interactions within the tumor microenvironment. Essentially, our characterization of this interplay is akin to a phenotypic description rather than a deciphering of the mechanistic networks that link LPA signaling to downstream effects that mediate tumor biology. Decoding these pathways and their modulators will require insights from multiple omics platforms with subsequent testing and validation in realistic but manipulable models and simulators of tumor dynamics.
Fundamentally, we still do not understand how ATX/LPA signaling that mediates inflammatory processes for physiological wound healing purposes is subverted into mediating chronic inflammatory disease states, which in some cases can lead to and propagate tumorigenesis. It is likely LPA signaling is a dual-edged sword to the host organism in cancer initiation. For example, in situations where cancer initiation is highly associated with chronic inflammation, such as inflammatory bowel disease in colorectal cancer, hepatitis in hepatocellular carcinoma or obesity-related low-grade inflammation in breast cancer, LPA signaling as a mediator of upregulated inflammatory cytokine production acts from the onset as a central orchestrator of the inflammation tropism central to the hallmarks of tumorigenesis in these malignancies (Fig. 3). However, in cases where tumorigenesis initiation is primarily mediated by other cancer hallmarks, such as genomic instabilities, LPA signaling enrichment derived from upregulated ATX expression in the stroma of the early tumor microenvironment may still retain its wound healing phenotype. This could explain why in early human breast cancers increased ATX was associated with better patient survival metrics [40], but the opposite in more advanced disease stages [27, 28]. However, through a combination of inflammatory signaling dysregulation and immune system evasion, breast cancers and other malignancies are able to temporally subvert LPA signaling to become maladaptive. This is ultimately responsible for driving tumor growth, metastasis, and resistance to therapeutic interventions (Fig. 3).
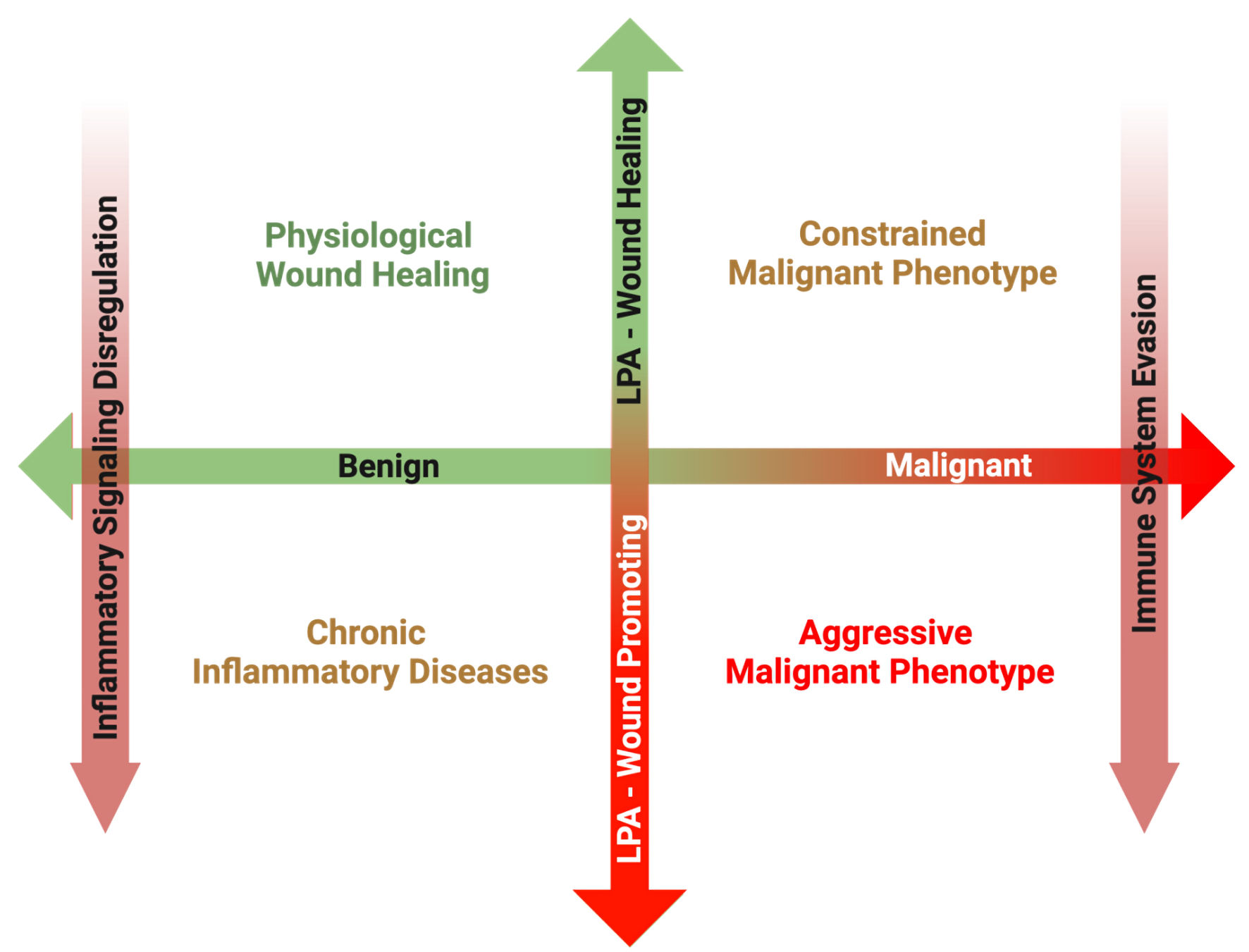 Click for large image | Figure 3. Overview of the proposed matrix between disease transformation and LPA subversion from a physiological wound healing mediator to a pathological wound promoting ligand. In the post-natal organism, a primary role of LPA signaling is to promote physiological wound healing processes in response to acute inflammation following tissue damage. Once the tissue is repaired, inflammation subsides, and increased LPA signaling is returned to basal levels. However, in the case of chronic inflammatory conditions, inflammatory pathways remain upregulated, resulting in increased ATX production and sustained LPA signaling. Some cancers can arise secondary to the sequalae of chronic inflammation, and in these cases, LPA signaling from tumor initiation perpetuates the chronicity of sustained inflammation, promoting tumorigenesis. However, during the early phases of some cancer initiation, LPA signaling from the surrounding host tissue can attempt to suppress development through its physiological wound healing mechanisms. In some cases, this LPA signaling is hijacked, and through a combination of inflammatory signaling dysfunction and immune system evasion, LPA signaling begins to mediate a pro-growth and pro-survival tumor microenvironment. LPA: lysophosphatidate; ATX: autotaxin. |
Now that inhibitors against LPA signaling have entered clinical trials, and ATX inhibitors are now in phase I trials for metastatic pancreatic cancer [63, 95], it should only be a matter of time before these inhibitors enter trials for breast cancer patients. While the ultimate goal of LPA signaling inhibition is to disrupt communication between the tumor microenvironment and cancer cells, and therapeutically mitigate tumor growth, metastasis, and the development of treatment resistance, further research is needed to determine the setting in which LPA inhibition would be most beneficial. Despite the fact that high ATX expression in early breast tumors appears to mitigate tumor growth, these same cancers also have increased expression of gene sets related to tumor stemness and treatment resistance, suggesting that these tumors maintain the propensity to subvert LPA signaling for nefarious purposes [40]. Under what conditions these cancers temporally hijack LPA for tumor propagation will be an ongoing area of investigation. Determining which tumors, and when they might benefit the most for LPA pathway signaling inhibition will likely be determined in part through applications of an LPA pathway-related gene signature to patient tumor characterization.
In addition to ongoing development and deducing the application of ATX inhibitors in breast cancer, targeting the LPA pathway via LPAR inhibition is likely to be most effective via inhibition of either LPAR2, given its high tumorigenic properties [15, 57], or LPAR5 to minimize its mediation of immune system evasion [59, 61]. Such inhibitors, however, may not have the intended therapeutic potency if the other LPARs can adequately compensate following their blockage, but further investigation in animal models will be required to determine this. If this were to be the case, a combination compound in the form of a dual ATX-inhibitor and selective LPAR inhibitor might be able to provide robust adjuvant therapy, as has been demonstrated with dual ATX-LPAR1 inhibition in metastatic melanoma models [86, 106]. Finally, pharmacological targeting of the LPPs remains an essentially untapped area of investigation. To date, there are no known selective LPP2 inhibitors. Similar to the argument for the LPARs, combination treatment with an ATX inhibitor might have synergistic effects by both depriving the tumor microenvironment of LPA, and additionally impeding cell cycling through blockade of LPP2-mediated cell cycling gene sets [41, 74].
In summary, over 30 years of basic science and recent translational investigations have started to unravel the intricacies of LPA signaling in the breast tumor microenvironment, and multiple pharmacological inhibitors are currently being investigated to treat a host of inflammatory-related diseases, including cancers. The ability to rationally design therapies against the LPA signaling pathway presents opportunities to target this signaling cascade both broadly and selectively at multiple levels. This affords the opportunity to potentially design treatments that may work as adjuncts to improve treatment effectiveness, either through increasing the therapeutic index of existing regimens, or improving drug bioavailability by blocking resistance mechanisms. Overall, LPA pathway inhibition represents an attractive strategy for improving patient care, not only for breast cancer, but multiple pathological conditions.
Acknowledgments
None to declare.
Financial Disclosure
This research was supported by the National Institutes of Health, USA grant number R37CA248018, R01CA250412, R01CA251545, R01EB029596, the US Department of Defense BCRP grant numbers W81XWH-19-1-0674 and W81XWH-19-1-0111 to K.T., and the National Cancer Institute Cancer Center Support Grant P30CA016056 to Roswell Park Comprehensive Cancer Center.
Conflict of Interest
The authors report no proprietary or commercial interest in any product mentioned or concept discussed in this article. D.N.B. is a member of the Scientific and Clinical Advisory Boards of iOnctura and receives funding for a postdoctoral fellow from iOnctura to work on an ATX inhibitor, but he has no commercial interests in the company. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Author Contributions
Conceptualization: M.G.K.B., D.N.B., K.T.; methodology: M.G.K.B.; data curation: M.G.K.B., X.T.; writing - original draft preparation: M.G.K.B.; writing - review and editing: X.T., D.N.B., K.T.; supervision: D.N.B., K.T.; project administration: M.G.K.B.; funding acquisition, K.T. All authors have read and agreed to the published version of the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
Abbreviations
ATX: autotaxin; IPF: idiopathic pulmonary fibrosis; LPA: lysophosphatidate; LPAR: lysophosphatidate receptors; LPC: lysophosphatidylcholine; LPP: lipid phosphate phosphatase; MAG: monoacylglycerol; MMTV: mammary mouse tumor virus
| References | ▴Top |
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7-33.
doi pubmed - Wang R, Zhu Y, Liu X, Liao X, He J, Niu L. The Clinicopathological features and survival outcomes of patients with different metastatic sites in stage IV breast cancer. BMC Cancer. 2019;19(1):1091.
doi pubmed pmc - Tufail M, Cui J, Wu C. Breast cancer: molecular mechanisms of underlying resistance and therapeutic approaches. Am J Cancer Res. 2022;12(7):2920-2949.
pubmed pmc - Benesch MGK, Tang X, Brindley DN. Autotaxin and Breast Cancer: Towards Overcoming Treatment Barriers and Sequelae. Cancers (Basel). 2020;12(2):374.
doi pubmed pmc - Benesch MG, Ko YM, McMullen TP, Brindley DN. Autotaxin in the crosshairs: taking aim at cancer and other inflammatory conditions. FEBS Lett. 2014;588(16):2712-2727.
doi pubmed - Fulkerson Z, Wu T, Sunkara M, Kooi CV, Morris AJ, Smyth SS. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J Biol Chem. 2011;286(40):34654-34663.
doi pubmed pmc - Balijepalli P, Sitton CC, Meier KE. Lysophosphatidic Acid Signaling in Cancer Cells: What Makes LPA So Special? Cells. 2021;10(8):2059.
doi pubmed pmc - Tang X, Benesch MG, Brindley DN. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J Lipid Res. 2015;56(11):2048-2060.
doi pubmed pmc - Stracke ML, Krutzsch HC, Unsworth EJ, Arestad A, Cioce V, Schiffmann E, Liotta LA. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J Biol Chem. 1992;267(4):2524-2529.
pubmed - Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J Biol Chem. 2002;277(42):39436-39442.
doi pubmed - Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J Cell Biol. 2002;158(2):227-233.
doi pubmed pmc - Brindley DN, Lin FT, Tigyi GJ. Role of the autotaxin-lysophosphatidate axis in cancer resistance to chemotherapy and radiotherapy. Biochim Biophys Acta. 2013;1831(1):74-85.
doi pubmed pmc - Aikawa S, Hashimoto T, Kano K, Aoki J. Lysophosphatidic acid as a lipid mediator with multiple biological actions. J Biochem. 2015;157(2):81-89.
doi pubmed - Leblanc R, Peyruchaud O. New insights into the autotaxin/LPA axis in cancer development and metastasis. Exp Cell Res. 2015;333(2):183-189.
doi pubmed - Benesch MGK, Wu R, Tang X, Brindley DN, Ishikawa T, Takabe K. Lysophosphatidic acid receptor signaling in the human breast cancer tumor microenvironment elicits receptor-dependent effects on tumor progression. Int J Mol Sci. 2023;24(12):9812.
doi pubmed pmc - Euer N, Schwirzke M, Evtimova V, Burtscher H, Jarsch M, Tarin D, Weidle UH. Identification of genes associated with metastasis of mammary carcinoma in metastatic versus non-metastatic cell lines. Anticancer Res. 2002;22(2A):733-740.
pubmed - Castellana B, Escuin D, Peiro G, Garcia-Valdecasas B, Vazquez T, Pons C, Perez-Olabarria M, et al. ASPN and GJB2 are implicated in the mechanisms of invasion of ductal breast carcinomas. J Cancer. 2012;3:175-183.
doi pubmed pmc - Hemmings DG, Brindley DN. Signalling by lysophosphatidate and its health implications. Essays Biochem. 2020;64(3):547-563.
doi pubmed - She S, Zhang Q, Shi J, Yang F, Dai K. Roles of autotaxin/autotaxin-lysophosphatidic acid axis in the initiation and progression of liver cancer. Front Oncol. 2022;12:922945.
doi pubmed pmc - Benesch MGK, MacIntyre ITK, McMullen TPW, Brindley DN. Coming of age for autotaxin and lysophosphatidate signaling: clinical applications for preventing, detecting and targeting tumor-promoting inflammation. Cancers (Basel). 2018;10(3):73.
doi pubmed pmc - Benesch MG, Tang X, Dewald J, Dong WF, Mackey JR, Hemmings DG, McMullen TP, et al. Tumor-induced inflammation in mammary adipose tissue stimulates a vicious cycle of autotaxin expression and breast cancer progression. FASEB J. 2015;29(9):3990-4000.
doi pubmed - Benesch MG, Tang X, Maeda T, Ohhata A, Zhao YY, Kok BP, Dewald J, et al. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J. 2014;28(6):2655-2666.
doi pubmed - Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, Yu S, et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell. 2009;15(6):539-550.
doi pubmed pmc - Benesch MG, Tang X, Venkatraman G, Bekele RT, Brindley DN. Recent advances in targeting the autotaxin-lysophosphatidate-lipid phosphate phosphatase axis in vivo. J Biomed Res. 2016;30(4):272-284.
doi pubmed pmc - Benesch MGK, Yang Z, Tang X, Meng G, Brindley DN. Lysophosphatidate Signaling: The Tumor Microenvironment's New Nemesis. Trends Cancer. 2017;3(11):748-752.
doi pubmed - Tang X, Morris AJ, Deken MA, Brindley DN. Autotaxin inhibition with IOA-289 decreases breast tumor growth in mice whereas knockout of autotaxin in adipocytes does not. Cancers (Basel). 2023;15(11):2937.
doi pubmed pmc - Popnikolov NK, Dalwadi BH, Thomas JD, Johannes GJ, Imagawa WT. Association of autotaxin and lysophosphatidic acid receptor 3 with aggressiveness of human breast carcinoma. Tumour Biol. 2012;33(6):2237-2243.
doi pubmed - Shao Y, Yu Y, He Y, Chen Q, Liu H. Serum ATX as a novel biomarker for breast cancer. Medicine (Baltimore). 2019;98(13):e14973.
doi pubmed pmc - Benesch MG, Zhao YY, Curtis JM, McMullen TP, Brindley DN. Regulation of autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J Lipid Res. 2015;56(6):1134-1144.
doi pubmed pmc - Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650-1659.
doi pubmed - Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9(8):628-638.
doi pubmed - Nakanaga K, Hama K, Aoki J. Autotaxin—an LPA producing enzyme with diverse functions. J Biochem. 2010;148(1):13-24.
doi pubmed - Kano K, Matsumoto H, Kono N, Kurano M, Yatomi Y, Aoki J. Suppressing postcollection lysophosphatidic acid metabolism improves the precision of plasma LPA quantification. J Lipid Res. 2021;62:100029.
doi pubmed pmc - Geraldo LHM, Spohr T, Amaral RFD, Fonseca A, Garcia C, Mendes FA, Freitas C, et al. Role of lysophosphatidic acid and its receptors in health and disease: novel therapeutic strategies. Signal Transduct Target Ther. 2021;6(1):45.
doi pubmed pmc - Volden PA, Skor MN, Johnson MB, Singh P, Patel FN, McClintock MK, Brady MJ, et al. Mammary adipose tissue-derived lysophospholipids promote estrogen receptor-negative mammary epithelial cell proliferation. Cancer Prev Res (Phila). 2016;9(5):367-378.
doi pubmed pmc - Schmid R, Wolf K, Robering JW, Strauss S, Strissel PL, Strick R, Rubner M, et al. ADSCs and adipocytes are the main producers in the autotaxin-lysophosphatidic acid axis of breast cancer and healthy mammary tissue in vitro. BMC Cancer. 2018;18(1):1273.
doi pubmed pmc - Dusaulcy R, Rancoule C, Gres S, Wanecq E, Colom A, Guigne C, van Meeteren LA, et al. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J Lipid Res. 2011;52(6):1247-1255.
doi pubmed pmc - Rancoule C, Dusaulcy R, Treguer K, Gres S, Guigne C, Quilliot D, Valet P, et al. Depot-specific regulation of autotaxin with obesity in human adipose tissue. J Physiol Biochem. 2012;68(4):635-644.
doi pubmed - Vandeweyer E, Hertens D. Quantification of glands and fat in breast tissue: an experimental determination. Ann Anat. 2002;184(2):181-184.
doi pubmed - Benesch MG, Wu R, Tang X, Brindley DN, Ishikawa T, Takabe K. Autotaxin production in the human breast cancer tumor microenvironment mitigates tumor progression in early breast cancers. Am J Cancer Res. 2023;13(7):2790-2813.
pubmed pmc - Benesch MGK, Wu R, Tang X, Brindley DN, Ishikawa T, Takabe K. Decreased lipid phosphate phosphatase 1/3 and increased lipid phosphate phosphatase 2 expression in the human breast cancer tumor microenvironment promotes tumor progression and immune system evasion. Cancers (Basel). 2023;15(8):2299.
doi pubmed pmc - Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, Thennavan A, et al. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet. 2021;53(9):1334-1347.
doi pubmed pmc - Koike S, Yutoh Y, Keino-Masu K, Noji S, Masu M, Ohuchi H. Autotaxin is required for the cranial neural tube closure and establishment of the midbrain-hindbrain boundary during mouse development. Dev Dyn. 2011;240(2):413-421.
doi pubmed - van Meeteren LA, Ruurs P, Stortelers C, Bouwman P, van Rooijen MA, Pradere JP, Pettit TR, et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol Cell Biol. 2006;26(13):5015-5022.
doi pubmed pmc - Turner JA, Fredrickson MA, D'Antonio M, Katsnelson E, MacBeth M, Van Gulick R, Chimed TS, et al. Lysophosphatidic acid modulates CD8 T cell immunosurveillance and metabolism to impair anti-tumor immunity. Nat Commun. 2023;14(1):3214.
doi pubmed pmc - Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479.
doi pubmed pmc - Panagopoulou M, Drosouni A, Fanidis D, Karaglani M, Balgkouranidou I, Xenidis N, Aidinis V, et al. ENPP2 promoter methylation correlates with decreased gene expression in breast cancer: implementation as a liquid biopsy biomarker. Int J Mol Sci. 2022;23(7):3717.
doi pubmed pmc - Ivan J, Patricia G, Agustriawan D. In silico study of cancer stage-specific DNA methylation pattern in White breast cancer patients based on TCGA dataset. Comput Biol Chem. 2021;92:107498.
doi pubmed - Brindley DN, Tang X, Meng G, Benesch MGK. Role of adipose tissue-derived autotaxin, lysophosphatidate signaling, and inflammation in the progression and treatment of breast cancer. Int J Mol Sci. 2020;21(16):5938.
doi pubmed pmc - Drosouni A, Panagopoulou M, Aidinis V, Chatzaki E. Autotaxin in breast cancer: role, epigenetic regulation and clinical implications. Cancers (Basel). 2022;14(21):5437.
doi pubmed pmc - Nowak MA, Boerlijst MC, Cooke J, Smith JM. Evolution of genetic redundancy. Nature. 1997;388(6638):167-171.
doi pubmed - Yung YC, Stoddard NC, Chun J. LPA receptor signaling: pharmacology, physiology, and pathophysiology. J Lipid Res. 2014;55(7):1192-1214.
doi pubmed pmc - Chun J, Hla T, Lynch KR, Spiegel S, Moolenaar WH. International Union of Basic and Clinical Pharmacology. LXXVIII. Lysophospholipid receptor nomenclature. Pharmacol Rev. 2010;62(4):579-587.
doi pubmed pmc - Spencer SA, Suarez-Pozos E, Soto-Verdugo J, Wang H, Afshari FS, Li G, Manam S, et al. Lysophosphatidic acid signaling via LPA(6) : A negative modulator of developmental oligodendrocyte maturation. J Neurochem. 2022;163(6):478-499.
doi pubmed pmc - Contos JJ, Ishii I, Fukushima N, Kingsbury MA, Ye X, Kawamura S, Brown JH, et al. Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2). Mol Cell Biol. 2002;22(19):6921-6929.
doi pubmed pmc - Yasuda D, Kobayashi D, Akahoshi N, Ohto-Nakanishi T, Yoshioka K, Takuwa Y, Mizuno S, et al. Lysophosphatidic acid-induced YAP/TAZ activation promotes developmental angiogenesis by repressing Notch ligand Dll4. J Clin Invest. 2019;129(10):4332-4349.
doi pubmed pmc - Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, Zhang Y, et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013;73(11):3470-3480.
doi pubmed pmc - Slaney CY, Kershaw MH, Darcy PK. Trafficking of T cells into tumors. Cancer Res. 2014;74(24):7168-7174.
doi pubmed - Mathew D, Kremer KN, Strauch P, Tigyi G, Pelanda R, Torres RM. LPA(5) is an inhibitory receptor that suppresses CD8 T-cell cytotoxic function via disruption of early TCR signaling. Front Immunol. 2019;10:1159.
doi pubmed pmc - Aiello S, Casiraghi F. Lysophosphatidic acid: promoter of cancer progression and of tumor microenvironment development. A promising target for anticancer therapies? Cells. 2021;10(6):1390.
doi pubmed pmc - Oda SK, Strauch P, Fujiwara Y, Al-Shami A, Oravecz T, Tigyi G, Pelanda R, et al. Lysophosphatidic acid inhibits CD8 T cell activation and control of tumor progression. Cancer Immunol Res. 2013;1(4):245-255.
doi pubmed pmc - Cortes J, Rugo HS, Cescon DW, Im SA, Yusof MM, Gallardo C, Lipatov O, et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. N Engl J Med. 2022;387(3):217-226.
doi pubmed - Deken MA, Niewola-Staszkowska K, Peyruchaud O, Mikulcic N, Antolic M, Shah P, Cheasty A, et al. Characterization and translational development of IOA-289, a novel autotaxin inhibitor for the treatment of solid tumors. Immunooncol Technol. 2023;18:100384.
doi pubmed pmc - Lei J, Guo S, Li K, Tian J, Zong B, Ai T, Peng Y, et al. Lysophosphatidic acid receptor 6 regulated by miR-27a-3p attenuates tumor proliferation in breast cancer. Clin Transl Oncol. 2022;24(3):503-516.
doi pubmed pmc - Tao K, Guo S, Chen R, Yang C, Jian L, Yu H, Liu S. Lysophosphatidic acid receptor 6 (LPAR6) expression and prospective signaling pathway analysis in breast cancer. Mol Diagn Ther. 2019;23(1):127-138.
doi pubmed - Lin YH, Lin YC, Chen CC. Lysophosphatidic Acid Receptor Antagonists and Cancer: The Current Trends, Clinical Implications, and Trials. Cells. 2021;10(7):1629.
doi pubmed pmc - Takahashi K, Fukushima K, Onishi Y, Inui K, Node Y, Fukushima N, Honoki K, et al. Lysophosphatidic acid (LPA) signaling via LPA(4) and LPA(6) negatively regulates cell motile activities of colon cancer cells. Biochem Biophys Res Commun. 2017;483(1):652-657.
doi pubmed - Morris AJ, Smyth SS. Lipid phosphate phosphatases: more than one way to put the brakes on LPA signaling? J Lipid Res. 2014;55(11):2195-2197.
doi pubmed pmc - Tang X, Benesch MG, Dewald J, Zhao YY, Patwardhan N, Santos WL, Curtis JM, et al. Lipid phosphate phosphatase-1 expression in cancer cells attenuates tumor growth and metastasis in mice. J Lipid Res. 2014;55(11):2389-2400.
doi pubmed pmc - Nakayama J, Raines TA, Lynch KR, Slack-Davis JK. Decreased peritoneal ovarian cancer growth in mice lacking expression of lipid phosphate phosphohydrolase 1. PLoS One. 2015;10(3):e0120071.
doi pubmed pmc - Tang X, McMullen TPW, Brindley DN. Increasing the low lipid phosphate phosphatase 1 activity in breast cancer cells decreases transcription by AP-1 and expressions of matrix metalloproteinases and cyclin D1/D3. Theranostics. 2019;9(21):6129-6142.
doi pubmed pmc - Flanagan JM, Funes JM, Henderson S, Wild L, Carey N, Boshoff C. Genomics screen in transformed stem cells reveals RNASEH2A, PPAP2C, and ADARB1 as putative anticancer drug targets. Mol Cancer Ther. 2009;8(1):249-260.
doi pubmed - Morris KE, Schang LM, Brindley DN. Lipid phosphate phosphatase-2 activity regulates S-phase entry of the cell cycle in Rat2 fibroblasts. J Biol Chem. 2006;281(14):9297-9306.
doi pubmed - Tang X, Cromwell CR, Liu R, Godbout R, Hubbard BP, McMullen TPW, Brindley DN. Lipid phosphate phosphatase-2 promotes tumor growth through increased c-Myc expression. Theranostics. 2022;12(13):5675-5690.
doi pubmed pmc - Venkatraman G, Benesch MG, Tang X, Dewald J, McMullen TP, Brindley DN. Lysophosphatidate signaling stabilizes Nrf2 and increases the expression of genes involved in drug resistance and oxidative stress responses: implications for cancer treatment. FASEB J. 2015;29(3):772-785.
doi pubmed - Iwaki Y, Ohhata A, Nakatani S, Hisaichi K, Okabe Y, Hiramatsu A, Watanabe T, et al. ONO-8430506: a novel autotaxin inhibitor that enhances the antitumor effect of paclitaxel in a breast cancer model. ACS Med Chem Lett. 2020;11(6):1335-1341.
doi pubmed pmc - Tang X, Wuest M, Benesch MGK, Dufour J, Zhao Y, Curtis JM, Monjardet A, et al. Inhibition of autotaxin with GLPG1690 increases the efficacy of radiotherapy and chemotherapy in a mouse model of breast cancer. Mol Cancer Ther. 2020;19(1):63-74.
doi pubmed - Maher TM, Ford P, Brown KK, Costabel U, Cottin V, Danoff SK, Groenveld I, et al. Ziritaxestat, a novel autotaxin inhibitor, and lung function in idiopathic pulmonary fibrosis: the ISABELA 1 and 2 randomized clinical trials. JAMA. 2023;329(18):1567-1578.
doi pubmed pmc - Maher TM, Kreuter M, Lederer DJ, Brown KK, Wuyts W, Verbruggen N, Stutvoet S, et al. Rationale, design and objectives of two phase III, randomised, placebo-controlled studies of GLPG1690, a novel autotaxin inhibitor, in idiopathic pulmonary fibrosis (ISABELA 1 and 2). BMJ Open Respir Res. 2019;6(1):e000422.
doi pubmed pmc - Xu X, Yu W, Kim M, Kim D, Fuentes ME, Huang J-X, et al. Differentiating characteristics of cudetaxestat (BLD-0409), a non-competitive autotaxin inhibitor under development to treat idiopathic pulmonary fibrosis. D29 MECHANISMS IN LUNG INJURY, REPAIR, AND FIBROSIS. pp. A5234-A5234.
- ClinicalTrials.gov. RESPIRARE - efficacy and safety of cudetaxestat in patients with idiopathic pulmonary fibrosis (IPF). https://clinicaltrials.gov/study/NCT05373914: National Institute of Health, 2022.
- ClinicalTrials.gov. To evaluate the efficacy, safety, and tolerability of BBT-877 in patients with IPF. https://clinicaltrials.gov/study/NCT05483907: National Institute of Health. 2023.
- Zhang C, Liu Y, Zhou Q, Fan H, Liu X, Hu J. Recent research advances in ATX inhibitors: An overview of primary literature. Bioorg Med Chem. 2023;90:117374.
doi pubmed - Bhave SR, Dadey DY, Karvas RM, Ferraro DJ, Kotipatruni RP, Jaboin JJ, Hallahan AN, et al. Autotaxin inhibition with PF-8380 enhances the radiosensitivity of human and murine glioblastoma cell lines. Front Oncol. 2013;3:236.
doi pubmed pmc - St-Coeur PD, Ferguson D, Morin P, Jr., Touaibia M. PF-8380 and closely related analogs: synthesis and structure-activity relationship towards autotaxin inhibition and glioma cell viability. Arch Pharm (Weinheim). 2013;346(2):91-97.
doi pubmed - Banerjee S, Lee S, Norman DD, Tigyi GJ. Designing dual inhibitors of autotaxin-LPAR GPCR axis. Molecules. 2022;27(17):5487.
doi pubmed pmc - Stein AJ, Bain G, Prodanovich P, Santini AM, Darlington J, Stelzer NM, Sidhu RS, et al. Structural basis for inhibition of human autotaxin by four potent compounds with distinct modes of binding. Mol Pharmacol. 2015;88(6):982-992.
doi pubmed - Clark JM, Salgado-Polo F, Macdonald SJF, Barrett TN, Perrakis A, Jamieson C. Structure-based design of a novel class of autotaxin inhibitors based on endogenous allosteric modulators. J Med Chem. 2022;65(8):6338-6351.
doi pubmed pmc - Barbayianni E, Magrioti V, Moutevelis-Minakakis P, Kokotos G. Autotaxin inhibitors: a patent review. Expert Opin Ther Pat. 2013;23(9):1123-1132.
doi pubmed - Hoeglund AB, Howard AL, Wanjala IW, Pham TC, Parrill AL, Baker DL. Characterization of non-lipid autotaxin inhibitors. Bioorg Med Chem. 2010;18(2):769-776.
doi pubmed - Jia Y, Li Y, Xu XD, Tian Y, Shang H. Design and development of autotaxin inhibitors. Pharmaceuticals (Basel). 2021;14(11):1203.
doi pubmed pmc - Nikolaou A, Kokotou MG, Limnios D, Psarra A, Kokotos G. Autotaxin inhibitors: a patent review (2012-2016). Expert Opin Ther Pat. 2017;27(7):815-829.
doi pubmed - Tan Z, Lei H, Guo M, Chen Y, Zhai X. An updated patent review of autotaxin inhibitors (2017-present). Expert Opin Ther Pat. 2021;31(5):421-434.
doi pubmed - Centonze M, Di Conza G, Lahn M, Fabregat I, Dituri F, Gigante I, Serino G, et al. Autotaxin inhibitor IOA-289 reduces gastrointestinal cancer progression in preclinical models. J Exp Clin Cancer Res. 2023;42(1):197.
doi pubmed pmc - ClinicalTrials.gov. A study to assess an ATX inhibitor (IOA-289) in patients with metastatic pancreatic cancer. https://clinicaltrials.gov/ct2/show/NCT05586516: National Institute of Health. 2022.
- Yang L, Shu P, Wu N, Hu M, Luo Z. Pharmacokinetics, pharmacodynamics, safety and tolerability of FTP-198, a novel, selective Autotaxin inhibitor, in healthy subjects: A phase I randomized placebo-controlled trial. Eur J Pharm Sci. 2023;189:106552.
doi pubmed - Shinde AT, Singh XR, Brindley DN. Infliximab, a monoclonal antibody against TNF-α, inhibits NF-κB activation, autotaxin expression and breast cancer metastasis to lungs. Cancers. 2024;16(1):52.
- Corte TJ, Lancaster L, Swigris JJ, Maher TM, Goldin JG, Palmer SM, Suda T, et al. Phase 2 trial design of BMS-986278, a lysophosphatidic acid receptor 1 (LPA(1)) antagonist, in patients with idiopathic pulmonary fibrosis (IPF) or progressive fibrotic interstitial lung disease (PF-ILD). BMJ Open Respir Res. 2021;8(1):e001026.
doi pubmed pmc - Palmer SM, Snyder L, Todd JL, Soule B, Christian R, Anstrom K, Luo Y, et al. Randomized, double-blind, placebo-controlled, phase 2 trial of BMS-986020, a lysophosphatidic acid receptor antagonist for the treatment of idiopathic pulmonary fibrosis. Chest. 2018;154(5):1061-1069.
doi pubmed - Allanore Y, Distler O, Jagerschmidt A, Illiano S, Ledein L, Boitier E, Agueusop I, et al. Lysophosphatidic acid receptor 1 antagonist SAR100842 for patients with diffuse cutaneous systemic sclerosis: a double-blind, randomized, eight-week placebo-controlled study followed by a sixteen-week open-label extension study. Arthritis Rheumatol. 2018;70(10):1634-1643.
doi pubmed - Liu W, Hopkins AM, Hou J. The development of modulators for lysophosphatidic acid receptors: A comprehensive review. Bioorg Chem. 2021;117:105386.
doi pubmed - Gu Z, Yan Y, Yao H, Lin K, Li X. Targeting the LPA1 signaling pathway for fibrosis therapy: a patent review (2010-present). Expert Opin Ther Pat. 2022;32(10):1097-1122.
doi pubmed - Tang X, Brindley DN. Lipid phosphate phosphatases and cancer. Biomolecules. 2020;10(9):1263.
doi pubmed pmc - Tang X, Zhao YY, Dewald J, Curtis JM, Brindley DN. Tetracyclines increase lipid phosphate phosphatase expression on plasma membranes and turnover of plasma lysophosphatidate. J Lipid Res. 2016;57(4):597-606.
doi pubmed pmc - Meng G, Tang X, Yang Z, Zhao Y, Curtis JM, McMullen TPW, Brindley DN. Dexamethasone decreases the autotaxin-lysophosphatidate-inflammatory axis in adipose tissue: implications for the metabolic syndrome and breast cancer. FASEB J. 2019;33(2):1899-1910.
doi pubmed - Lee SC, Fujiwara Y, Liu J, Yue J, Shimizu Y, Norman DD, Wang Y, et al. Autotaxin and LPA1 and LPA5 receptors exert disparate functions in tumor cells versus the host tissue microenvironment in melanoma invasion and metastasis. Mol Cancer Res. 2015;13(1):174-185.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.