

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 14, Number 6, December 2023, pages 558-569

Genetically Predicted Causal Effects of Gut Microbiota and Gut Metabolites on Digestive Tract Cancer: A Two-Sample Mendelian Randomization Analysis
Xu Jia Lia, b, c, e, Meng Ge Gaod, e, Xu Xian Chena, b, c, Yu Ming Ronga, b, c, Ling Li Huanga, b, c, f, Jin Sheng Huanga, b, c, f ![]()
aVIP Department, Sun Yat-sen University Cancer Center, Guangzhou 510060, China
bState Key Laboratory of Oncology in South China, Sun Yat-sen University Cancer Center, Guangzhou 510060, China
cCollaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou 510060, China
dDepartment of Clinical Nutrition, Huadu District People’s Hospital, Southern Medical University, Guangzhou 510800, China
eThese authors contributed equally to this work.
fCorresponding Author: Jin Sheng Huang and Ling Li Huang, VIP Department, Sun Yat-sen University Cancer Center, Guangzhou 510060, Chinaand
Manuscript submitted September 27, 2023, accepted October 20, 2023, published online November 3, 2023
Short title: Gut Microbiota and Metabolites in Digestive Tract Cancer
doi: https://doi.org/10.14740/wjon1737
| Abstract | ▴Top |
Background: Evidence from numerous observational studies and clinical trials has linked gut microbiota and metabolites to digestive tract cancer. However, the causal effect between these factors remains uncertain.
Methods: Data for this study were obtained from the MiBioGen, TwinsUK Registry, and FinnGen (version R8). Two-sample Mendelian randomization analysis with inverse variance weighting method was primarily used, and the results were validated by heterogeneity analysis, pleiotropy test, and sensitivity analysis.
Results: At P < 5 × 10-8, our analysis identified four gut microbiotas as risk factors for digestive tract cancer and six as risk factors for colorectal cancer. Conversely, one gut microbiota exhibited protection against bile duct cancer, and two showed protective effects against stomach cancer. At P < 1 × 10-5, our investigation revealed five, six, three, eight, eight, and eight gut microbiotas as risk factors for esophageal, stomach, bile duct, liver, pancreatic, and colorectal cancers, respectively. In contrast, four, two, eight, two, two, and five gut microbiotas exhibited protective effects against these cancers. Additionally, GABA, a metabolite of gut microbiota, displayed a significant protective effect against colorectal cancer.
Conclusion: In conclusion, specific gut microbiota and metabolites play roles as risk factors or protective factors for digestive tract cancer, and a causal relationship between them has been established, offering novel insights into gut microbiota-mediated cancer development.
Keywords: Gut microbiota; Gut metabolites; Digestive tract cancer; Causal effect; Mendelian randomization
| Introduction | ▴Top |
The gut microbiota emerges as a pivotal determinant of host well-being, exerting its influence from the earliest stages of life [1, 2]. Among the diverse microbial communities inhabiting the human body, the gut microbiome has garnered considerable attention and extensive exploration [3]. Mounting evidence implicates intricate associations between cancer development and somatic mutations, epigenetic modifications in neoplastic cells, as well as interplay among host genetic variations, immune responses, environmental exposures, and the microbiome [4]. Leveraging cutting-edge molecular tools, researchers are progressively unraveling the intricate dynamics between the host and diverse microbial entities, wherein the gut microbiota has been recognized as a potentially influential risk or protective factor across a spectrum of diseases, including cancer [5, 6]. Despite the weighty epidemiological evidence, discerning the precise contribution of microbes to human cancer pathogenesis remains a formidable challenge, warranting meticulous inquiry into the underlying genetic and molecular mechanisms that underpin their putative causal relationship.
Over the past decade, genome-wide association studies (GWAS) have revolutionized the landscape of complex disease genetics by examining millions of genetic variants to uncover associations between genotypes and phenotypes [7]. By investigating the relationship between common single-nucleotide polymorphisms (SNPs) and diseases or other phenotypic traits on a genome-wide scale, GWAS have opened up novel avenues for comprehending the underlying mechanisms of complex diseases. Additionally, Mendelian randomization (MR) analyses employ genetic variants, typically SNPs, as instrumental variables (IVs) to infer potential causal relationships between exposures and outcomes [8-10]. Since SNPs are randomly allocated at the point of conception and remain unaffected by confounding variables, the impact of such confounding factors can be minimized. In addition, because the genotype of an individual’s exposure is determined at the time of conception, this cannot be changed, whereas the SNPs for the outcome are de-matched by exposure, meaning that the exposure precedes the outcome. Therefore, there will be no reverse causality where causality is associated with genotype, which is another advantage of MR. To summarize, MR analysis offers more robust evidence for inferring causality than traditional observational studies [10, 11].
Given the absence of studies investigating the causal link between gut microbiota, gut metabolites, and digestive tract cancer, we undertook a population-based study on a European cohort to examine this relationship. The objective of our study was to ascertain the existence of a causal relationship between gut microbiota, gut metabolites, and digestive tract cancer. The findings from this study will contribute to a deeper understanding of the involvement of gut microbiota and metabolites in the pathogenesis of digestive tract cancer, leading to the development of more targeted cancer surveillance protocols. These protocols aim to enable early detection of cancer or pre-cancerous lesions, thereby reducing the burden on healthcare resources.
| Materials and Methods | ▴Top |
A two-sample MR design was employed to investigate the causal relationship between gut microbiota, gut metabolites, and the risk of developing digestive tract cancer. In this MR study, we considered gut microbiota as the exposure factor and digestive tract cancer as the outcome. To fulfill the requirements of the MR approach, independent genetic variants were utilized as IVs, and they needed to satisfy three crucial assumptions [11]: 1) strong association of IVs with the exposure; 2) absence of pleiotropic associations between IVs and any known confounding factors; and 3) absence of prognostic associations, except potentially with the exposure. To ensure the integrity of the analysis, genetic data pertaining to gut microbiota, gut metabolites, and digestive tract cancer were extracted from separate GWAS datasets, thereby eliminating sample overlap. A comprehensive overview of this MR study can be found in Figure 1.
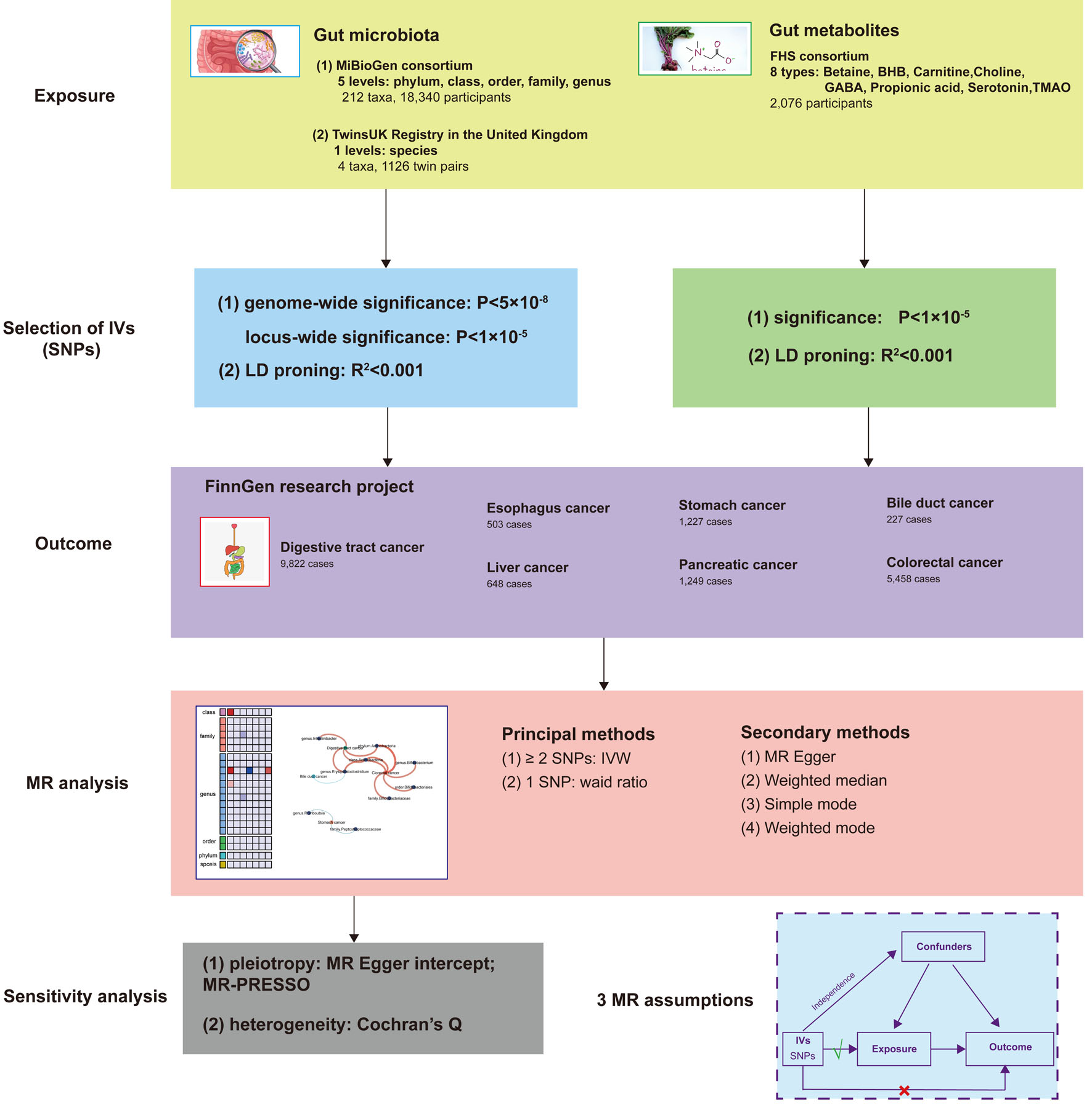 Click for large image | Figure 1. Study design of the two-sample MR for the effect of the genetically predicted gut microbiome and gut metabolites on digestive tract cancer. MiBioGen: international consortium MiBioGen; TwinsUK Registry: the UK adult twin registry; FHS: Framingham Heart Study; LD: linkage disequilibrium; SNP: single-nucleotide polymorphism; MR: Mendelian randomization; IVs: instrumental variables; IVW: inverse-variance weighted. |
Exposure data
Summary data on gut microbiota were collected from two sources: The MiBioGen (international consortium MiBioGen) and TwinsUK Registry (The UK Adult Twin Registry), which included five levels (phylum, class, order, family, and genus), 212 taxa, 18,340 participants and one level (species), four taxa, 1,126 twin pairs, respectively. Additionally, data on gut metabolites were derived from the Framingham Heart Study (FHS), encompassing eight types of metabolites (betaine, β-hydroxybutyric acid (BHB), carnitine, choline, γ-amino-butyric acid (GABA), propionic acid, serotonin, trimethylamine N-oxide (TMAO)), with a total of 2,076 participants included in the analysis. The specifics are shown in Table 1. No weak IVs were identified among the exposure factors, and all F-statistics exceeded 10, indicating minimal bias due to weak IVs (Supplementary Tables S1, S2, and S3, www.wjon.org). The MiBioGen dataset, a large multiethnic GWAS collaboration, consisted of 18,340 participants from 16 cohorts across various countries, including the United States, Canada, Israel, Korea, Germany, Denmark, the Netherlands, Belgium, Sweden, Finland, and the United Kingdom. This dataset included 24S ribosomal RNA gene sequencing and genotype data, enabling the exploration of the association between human autosomal gene variants and the gut microbiome [12]. The Twins UK Registry, established in 1993 at King’s College London, is the largest adult twin program in the UK. The registry comprises individuals ranging in age from 16 to 98 years and aims to investigate the genetic and environmental factors underlying complex traits and diseases [13]. FHS is the world’s largest population-based family study, characterized by a substantial sample size and a long observation period spanning three generations. The dataset is rich in cardiometabolic phenotype information and is based on family structure. It provides a unique opportunity to examine the impact of genetic, environmental, and clinical factors on the plasma metabolome [14, 15].
 Click to view | Table 1. Description of Gut Microbiota, Metabolites, and Digestive Tract Cancer |
IV selection
We conducted a comprehensive analysis of bacterial taxa at six levels (phylum, class, order, family, genus, and species), considering each unique taxon as a characteristic. To ensure the reliability of our conclusion regarding the causal relationship between gut microbiota, gut metabolites, and the risk of developing digestive tract cancer, we implemented a rigorous quality control process to select the most suitable IVs. First, we employed two thresholds to identify SNPs significantly associated with gut microbiota and gut metabolites as IVs. Using a genome-wide significance threshold of P < 5 × 10-8, only a limited number of IVs were available. To achieve a more comprehensive exploration of the potential causal relationship, we employed a lower genome-wide significance threshold of P < 1 × 10-5, resulting in the inclusion of additional IVs. Second, we applied a minor allele frequency (MAF) threshold of 0.01 for the variant of interest. Third, to mitigate bias, we selected exposure factors that were SNPs with no significant linkage disequilibrium (LD) and a closely pairwise correlation (R2 < 0.01), while considering a clumping distance of 10,000 kb. Fourth, we excluded palindromic SNPs (e.g., those with A/T or G/C alleles) to avoid potential issues related to strand orientation or allele coding. Additionally, we carefully compared the alleles with the human genome reference sequence (build 37) and removed ambiguous or duplicated SNPs. These stringent criteria were applied to ensure the validity and robustness of our analyses and to minimize potential biases in our results.
Outcome data
We utilized aggregated data from the FinnGen (R8) database that encompasses cancer-related GWAS studies. The pooled data consisted of information on digestive tract cancer, esophagus cancer, stomach cancer, bile duct cancer, liver cancer, pancreatic cancer, and colorectal cancer, with respective European population sizes of 9,822, 503, 1,227, 227, 648, 1,249, and 5,458 individuals (Table 1). The FinnGen database was established through a collaborative effort between academia and industry, with the aim of exploring genotype-phenotype correlations within the Finnish population and gaining insights into the impact of the genome on health [16].
MR analysis and sensitivity analysis
To explore potential causal relationships, we conducted a two-sample unidirectional MR study to investigate the association between gut microbiota, gut metabolites, and various digestive tract cancer, including esophagus cancer, stomach cancer, bile duct cancer, liver cancer, pancreatic cancer, and colorectal cancer. The primary analysis method used was the inverse-variance weighted (IVW) test [17], employing the IVW method when the number of SNPs was ≥ 2, and the weighted ratio method for analysis when only one SNP was available. Additional complementary methods, including weighted mode [18], MR-Egger intercept [19], weighted median [20], and simple mode [21], were also employed. The IVW method has been reported to exhibit slightly greater strength compared to other methods under specific conditions [20].
To ensure the robustness of the results, we conducted a sensitivity analysis. First, we employed MR-PRESSO [22] and MR-Egger intercept to assess potential horizontal pleiotropy. The MR-PRESSO test helped identify and exclude SNPs that might introduce bias, with a P-value > 0.05 indicating the absence of horizontal pleiotropy. The remaining SNPs were used for MR analysis. Deviation of the MR-Egger intercept from the origin suggested potential horizontal pleiotropy effects of the IVs, with a P-value < 0.05 indicating such effects, while a P-value ≥ 0.05 suggested no evidence of horizontal pleiotropy among the selected IVs. Second, we employed the weighted median as an additional sensitivity analysis to evaluate the robustness of the MR estimates. Third, we calculated the F-statistic to assess weak instrumental bias, considering a F-statistic < 10 as indicative of weak IVs that may introduce bias and should be excluded [23]. Furthermore, Cochran’s Q statistic was used to assess heterogeneity in the IVW model, with a Q value greater than the number of instruments minus 1, suggesting the presence of heterogeneity and invalid instruments. A P-value < 0.05 indicated the possible existence of heterogeneity [24].
All statistical analyses were performed using R (version 4.0.2) and the TwoSampleMR package (version 0.5.6), which are open-source software tools.
Ethics approval and consent to participate
Ethical approval was not required for this study because our analysis used publicly available GWAS summary data, and these original GWAS had previously been approved by the appropriate ethical and institutional review boards.
| Results | ▴Top |
SNP selection
After a quality control step, we identified 215 and 22 gut microbiota exposure phenotypes at the genus, family, order, class, phylum, and species levels of significance at P < 1 × 10-5 and P < 5 × 10-8, respectively, with a significance level of 131, 35, 20, 16, 9, 4 and 18, 5, 2, 1, 1, 2, respectively (Fig. 2a and Supplementary Figure S1A, www.wjon.org). At a significance level of P < 1 × 10-5, eight gut metabolites were finally identified as exposure phenotypes, namely betaine, BHB, carnitine, choline, GABA, propionic acid, serotonin, and TMAO (Fig. 3). The final number of IVs selected for each gut microbiota and gut microbiota metabolite is detailed in Supplementary Table S4 (www.wjon.org).
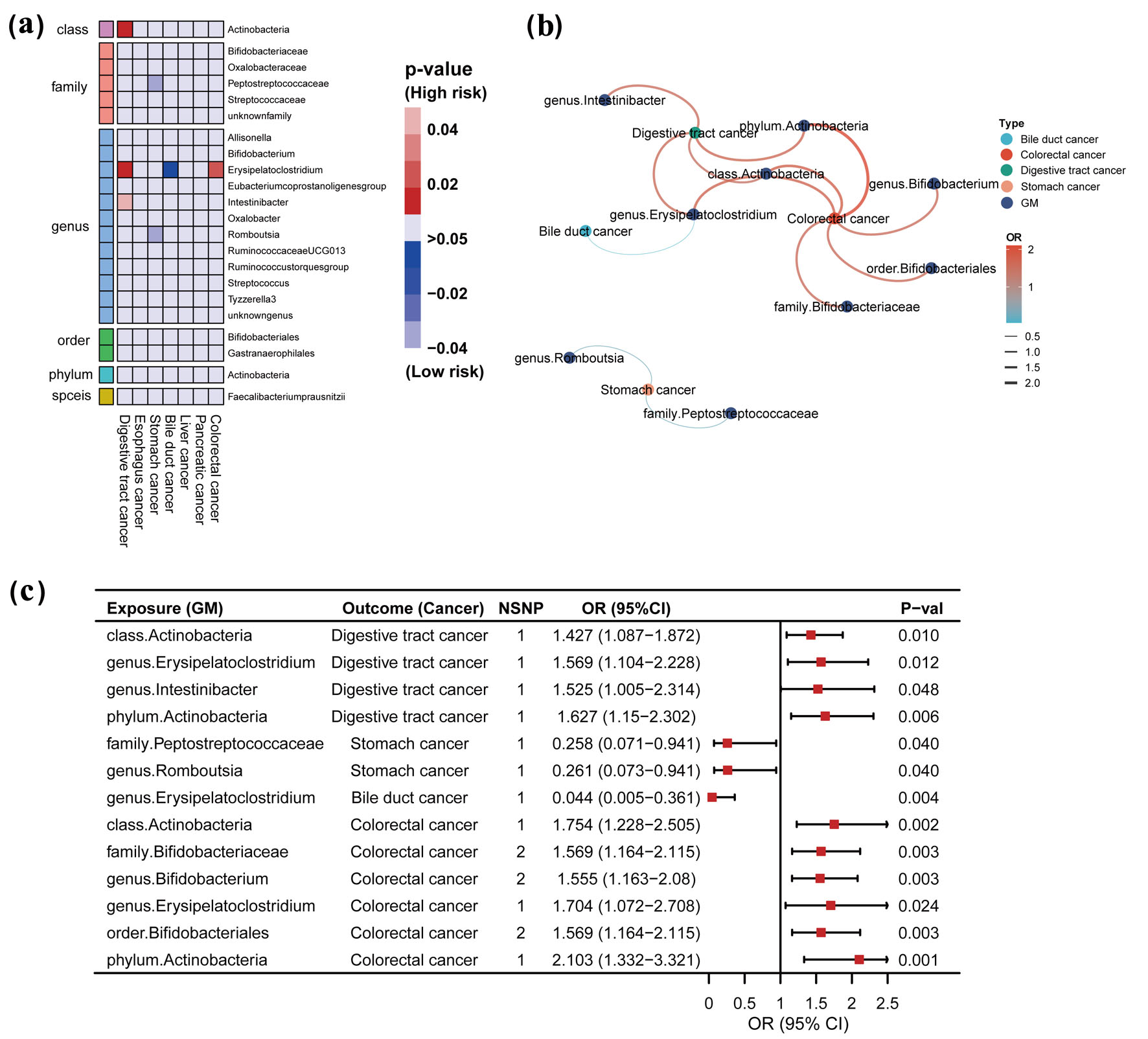 Click for large image | Figure 2. Causal effects of gut microbiome and digestive tract cancer based on the IVW method (SNPs with P < 5 × 10-8). (a) Heatmap of the P-value. Red color in the heatmap indicates a positive correlation between gut microbiome and digestive tract cancer and blue color indicates a negative correlation. The color depth represents the size of the P-value, with darker colors indicating more significant P-values. (b) Network interactions based on statistically significant ORs. Color and line thickness in the network diagram indicate the OR of gut microbiota and digestive tract cancer. Darker red color and thicker lines indicate higher OR values. (c) Forest plot with statistically significant ORs. OR > 1 indicates the positive correlation between a particular gut microbiota and a particular digestive tract cancer. OR < 1 indicates the negative correlation between the two. IVW: inverse-variance weighted; GM: gut microbiome; SNP: single-nucleotide polymorphism; OR: odds ratio; 95% CI: 95% confidence interval; NSNP: number of SNP. |
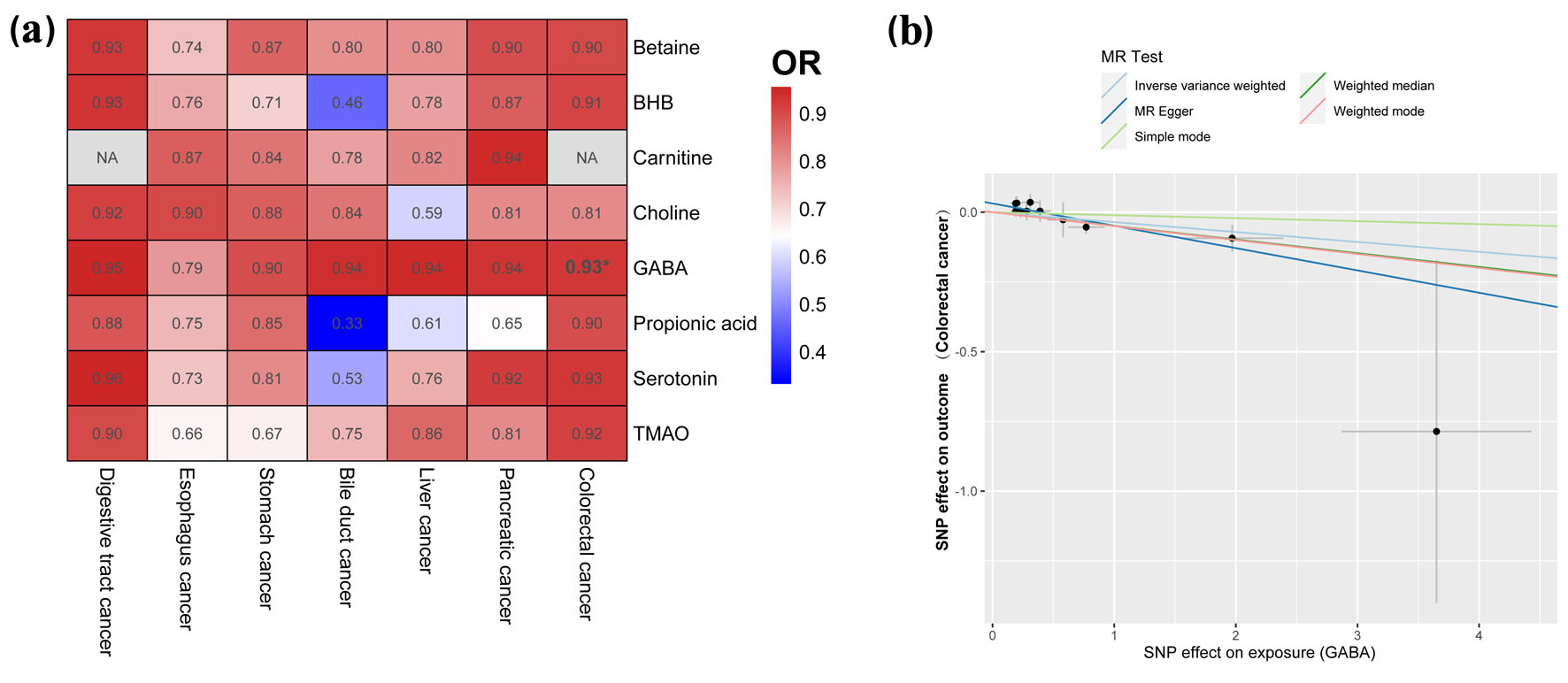 Click for large image | Figure 3. Causal effects of gut metabolite and digestive tract cancer based on the IVW method (SNPs with P < 1 × 10-5). (a) Heatmap of the ORs. Color in the heatmap indicates the OR of gut metabolite and digestive tract cancer. Darker red color indicates higher OR values. The asterisk indicates statistically significant OR. (b) Correlation between GABA and colorectal cancer. IVW: inverse-variance weighted; SNP: single-nucleotide polymorphism; OR: odds ratio; MR: Mendelian randomization. |
Causal relationship between gut microbiota and digestive tract cancer
Genome-wide statistical significance threshold P < 5 × 10-8
At a significance level of P < 5 × 10-8, our analysis revealed a causal relationship between specific gut microbiota and digestive tract cancer, including stomach, bile duct, and colorectal cancers (Fig. 2a, b). The IVW analyses indicated that certain microbial taxa, such as genus Erysipelatoclostridium (odds ratio (OR) = 1.569), class Actinobacteria (OR = 1.427), phylum Actinobacteria (OR = 1.627), and genus Intestinibacter (OR = 1.525), were associated with an increased risk of digestive tract cancer when considered as exposure factors. Similarly, for colorectal cancer, the presence of genus Erysipelatoclostridium (OR = 1.704), class Actinobacteria (OR= 1.754), family Bifidobacteriaceae (OR = 1.569), genus Bifidobacterium (OR = 1.555), order Bifidobacteriales (OR = 1.569), and phylum Actinobacteria (OR = 2.103) were associated with an increased risk. Interestingly, we also identified some protective factors, such as genus Erysipelatoclostridium (OR = 0.044) associated with a reduced risk of bile duct cancer, and family Peptostreptococcaceae (OR = 0.258) and genus Romboutsia (OR = 0.261) associated with a reduced risk of stomach cancer when used as exposure factors. For further details, refer to Figure 2c, Supplementary Tables S5 and S6 (www.wjon.org).
These results were statistically significant (P < 0.05), and the MR-Egger regression and Cochran’s Q test provided no evidence of bias or heterogeneity (Supplementary Table S7, www.wjon.org).
Genome-wide statistical significance threshold P < 1 × 10-5
At a significance level of P < 1 × 10-5, we observed a broader range of causal associations between gut microbiota and digestive tract cancer, including esophageal, stomach, bile duct, liver, pancreatic, and colorectal cancers. The IVW analyses revealed the following findings (Supplementary Tables S8 and S9, www.wjon.org).
For esophageal cancer, certain microbial taxa such as family ClostridialesvadinBB60group (OR = 2.164), genus unknowngenus.id.1000000073 (OR = 2.164), class Alphaproteobacteria (OR = 2.360), genus Coprococcus2 (OR = 2.396), and genus Sellimonas (OR = 1.438) were identified as risk factors, while family Lachnospiraceae (OR = 0.501), genus Butyricimonas (OR = 0.544), genus CandidatusSoleaferrea (OR = 0.532), and phylum Verrucomicrobia (OR = 0.5260) were protective factors (Supplementary Figure S1B and D, www.wjon.org).
For stomach cancer, genus Erysipelatoclostridium (OR = 1.540), genus Olsenella (OR = 1.257), genus Roseburia (OR = 1.589), genus RuminococcaceaeUCG014 (OR = 1.524), genus Streptococcus (OR = 1.641), and genus unknowngenus.id.959 (OR = 1.344) were associated with an increased risk, while genus LachnospiraceaeFCS020group (OR = 0.658) and genus RuminococcaceaeUCG004 (OR = 0.611) were protective factors (Supplementary Figure S1B and E, www.wjon.org).
For bile duct cancer, genus Alloprevotella (OR = 2.824), genus Tyzzerella3 (OR = 2.688), and order Lactobacillales (OR = 3.179) were identified as risk factors, while class Negativicutes (OR = 0.310), family unknownfamily.id.1000001214 (OR = 0.476), genus Anaerotruncus (OR = 0.3402), genus Howardella (OR = 0.574), genus unknowngenus.id.1000001215 (OR = 0.476), order Gastranaerophilales (OR = 0.476), order Selenomonadales (OR = 0.310), and species Akkermansiamuciniphila (OR = 0.743) were protective factors (Supplementary Figure S1B and F, www.wjon.org).
For liver cancer, genus Barnesiella (OR = 2.177), family Enterobacteriaceae (OR = 2.515), family unknownfamily.id.1000005471 (OR = 1.810), genus Oscillospira (OR = 2.121), genus unknowngenus.id.1000005472 (OR = 1.810), genus unknowngenus.id.2001 (OR = 1.742), order Enterobacteriales (OR = 2.515), and order MollicutesRF9 (OR = 1.810) were identified as risk factors, while genus Turicibacter (OR = 0.577) and phylum Firmicutes (OR = 0.570) were protective factors. Furthermore, among the risk factors identified for liver cancer, including family Enterobacteriaceae (OR = 1.288), family unknownfamily.id.1000005471 (OR = 1.159), genus unknowngenus.id.1000005472 (OR = 1.159), order Enterobacteriales (OR = 1.288), and order MollicutesRF9 (OR = 1.159), we observed that they were also associated with an increased risk of developing digestive tract cancer (Supplementary Figure S1B and G, www.wjon.org).
For pancreatic cancer, genus Flavonifractor (OR = 2.124), genus unknowngenus.id.2041 (OR = 1.555), class Erysipelotrichia (OR = 1.616), family Erysipelotrichaceae (OR = 1.616), genus Streptococcus (OR = 1.653), genus Terrisporobacter (OR = 1.671), genus Victivallis (OR = 1.258), and order Erysipelotrichales (OR = 1.616) were associated with an increased risk, while class Lentisphaeria (OR = 0.697) and order Victivallales (OR = 0.697) were associated with a decreased risk. Within the identified risk factors, we observed that genus Terrisporobacter (OR = 1.326) and genus Victivallis (OR = 1.103) were specifically associated with an increased risk of developing digestive tract cancer (Supplementary Figure S1B and H, www.wjon.org).
For colorectal cancer, family Porphyromonadaceae (OR = 1.699), class Coriobacteriia (OR = 1.319), family Coriobacteriaceae (OR = 1.319), family Enterobacteriaceae (OR = 1.494), family Lactobacillaceae (OR = 1.346), genus Sellimonas (OR = 1.142), order Coriobacteriales (OR = 1.319), and order Enterobacteriales (OR = 1.494) were associated with an increased risk, while class Bacteroidia (OR = 0.766), genus Eubacteriumfissicatenagroup (OR = 0.854), order Bacteroidales (OR = 0.766), phylum Bacteroidetes (OR = 0.680), and phylum Cyanobacteria (OR = 0.820) were protective factors. Among the identified factors, we observed that family Coriobacteriaceae (OR = 1.209), family Enterobacteriaceae (OR = 1.288), family Lactobacillaceae (OR = 1.213), family Porphyromonadaceae (OR = 1.389), genus Sellimonas (OR = 1.103), and order Coriobacteriales (OR = 1.209) were all associated with an increased risk of digestive tract cancer. On the other hand, phylum Bacteroidetes (OR = 0.790) was associated with a reduced risk of developing digestive tract cancer (Supplementary Figure S1B and I, www.wjon.org).
All these results showed statistical significance (P < 0.05). The MR-Egger regression and Cochran’s Q test indicated no evidence of bias or heterogeneous associations (Supplementary Table S10, www.wjon.org).
Causal relationship between gut metabolites and digestive tract cancer
In our analysis of the causal relationship between gut metabolites and digestive tract cancer, we conducted an IVW analysis on eight specific metabolites (at a significance level of P < 1 × 10-5): betaine, BHB, carnitine, choline, GABA, propionic acid, serotonin, and TMAO. Among these metabolites, our findings revealed that GABA, a metabolite produced by the gut microbiota, exhibited a protective effect against colorectal cancer (OR = 0.965) (Fig. 3, Supplementary Tables S11 and S12, www.wjon.org). The P-values obtained from the MR-Egger regression and Cochran’s Q test were both greater than 0.05, indicating no evidence of bias or heterogeneous associations (Supplementary Table S13, www.wjon.org).
Sensitivity analysis
To ensure the robustness of our MR causal effect estimates, we performed several sensitivity analyses. These included the utilization of MR-Egger, weighted mode, simple mode, and weighted median estimator (WME) methods. Across all analyses, no evidence of a horizontal pleiotropic effect was detected, as indicated by P-values greater than 0.05. Furthermore, no outliers were identified in the MR-PRESSO analyses, and the Cochran’s Q test revealed no significant heterogeneity (P > 0.05) (Supplementary Table S14, www.wjon.org).
| Discussion | ▴Top |
Our study utilized genetic variants from the largest intestinal microflora GWAS, focusing on those that exhibited strong associations with comprehensive genetic data. Through our investigation, we identified several gut microbiota and metabolites that potentially serve as risk or protective factors for digestive tract cancer, establishing a causal relationship between them. These findings hold significant implications for public health interventions aimed at reducing the risk of cancer. Notably, our study represents the first of its kind in exploring the causal relationship between gut microbiota, gut metabolites, and digestive tract cancer using MR analysis, as no prior studies have been published on this subject.
An increasing body of research has revealed a potential causal link between the gut microbiota we have selected and various digestive tract cancer, including esophageal, pancreatic, stomach, bile duct, liver, and colorectal cancers [25-30]. Additionally, several other studies have demonstrated associations between metabolites produced by the gut microbiota and the risk of developing digestive tract cancer [31-34].
In a specific study, 15S rRNA gene sequencing was utilized to compare the fecal microbiota composition of 16 patients diagnosed with esophageal squamous cell carcinoma (ESCC) and 16 healthy individuals serving as control subjects. The findings of this study indicated that the gut microbiota of ESCC patients exhibited potential enrichment in pro-inflammatory and/or oncogenic bacteria (e.g., Butyricimonas, Veillonella, and Streptococcus), while simultaneously showing depletion of butyrate-producing and/or potentially anti-inflammatory bacteria (e.g., Butyricicoccus, Lachnospiraceae NK4A136 group, and Eubacterium eligens group). Moreover, investigations have identified logarithmic ratios between Streptococcus and Butyricicoccus, as well as Streptococcus and Lachnospiraceae NK4A136 group, as potential diagnostic biomarkers for ESCC [35]. However, it is important to note that our study’s results suggest a potentially different role for Butyricimonas and Lachnospiraceae as protective factors against esophageal cancer. Further studies are required to validate these findings and provide more conclusive evidence.
Studies have consistently observed an elevated presence of Streptococcus in the intestines of patients diagnosed with stomach cancer [36-40]. Furthermore, investigations examining postoperative specimens of stomach cancer have reported an increased abundance of Streptococcus in stomach cancer tissues, suggesting its potential involvement as a causative agent in stomach cancer [41-43]. In alignment with these findings, our study also identified an increased risk of stomach cancer when Streptococcus was considered as an exposure factor. Additionally, another study noted that patients with gastric intraepithelial neoplasia exhibited an enrichment of specific intestinal commensals in their gastric microbiota, including Romboutsia, Fusicatenibacter, Prevotellaceae-Ga6A1-group, and Intestinimonas, which demonstrated a protective effect [44]. Consistent with these observations, our study revealed a reduced risk of stomach cancer when genus Romboutsia was utilized as an exposure factor.
Our study supports the finding that Lactobacillales is a risk factor for bile duct cancer. Similarly, a study conducted by Jia et al [45] investigated the fecal microorganisms of patients with intrahepatic bile duct cancer and observed increased levels of Lactobacillus, Actinomyces, Peptostreptococcaceae, and Alloscardovia compared to the healthy population. This discovery holds significant implications for the diagnosis and prediction of intrahepatic bile duct cancer.
The role of gut microbiota in the development and progression of liver cancer as a coordinator of the gut-liver axis has been emphasized [46]. Jiang et al [47] conducted a study investigating the gut microbiota of hepatocellular carcinoma (HCC) patients and identified higher abundances of Barnesiella, Firmicutes, and Streptococcus in their intestines. Similarly, Yang et al [48] observed an association between HCC and gut microbiota, with a higher abundance of Streptococcus in the gut of HCC patients. Consistent with these studies, our findings also indicate that Streptococcus and Barnesiella are risk factors for liver cancer. However, in contrast to previous findings, we found that Firmicutes are protective factors for liver cancer.
In our study, we identified Flavonifractor and Streptococcus as risk factors for pancreatic cancer. These findings are consistent with previous research. A large-scale metabolome-wide association study (MWAS) demonstrated an association between Flavonifractor sp90199495 and the metabolite X-21849, which was found to be related to the risk of pancreatic ductal adenocarcinoma [49]. Additionally, another study observed significant differences in gut microbial composition between pancreatic cancer patients and healthy individuals, with higher levels of Streptococcus being associated with an increased risk of pancreatic cancer development and liver metastasis. These findings suggest that Streptococcus may serve as a potential biomarker for the early diagnosis of pancreatic cancer and liver metastasis originating from pancreatic cancer [50]. Furthermore, Ogrendik’s study revealed a positive correlation between salivary microbiota and the risk of pancreatic cancer [51]. The research specifically identified Porphyromonas gingivalis as a significant risk factor for pancreatic cancer.
The gut microbiota exerts a crucial influence on the initiation and progression of colorectal cancer [52]. Certain researchers delve into the microbial mechanisms connected to the pathogenesis and advancement of colorectal cancer [53]. A meta-analytic study demonstrated a link between colorectal cancer and microbiota dysbiosis, with increased abundance of Porphyromonadaceae and Coriobacteriaceae in the intestines of colorectal cancer patients [54]. Another study conducted by Huo et al [55] investigated the gut mucosal microbiota associated with recurrence and survival in colorectal cancer patients. They observed that Bacteroidales, Coriobacteriaceae, and Porphyromonadaceae were associated with poorer survival and higher recurrence rates. Interestingly, they found that the effects of Bacteroidales differed based on their abundance at different tumor sites. High abundance of Bacteroidales at the extratumoral site was associated with better overall survival (OS) and disease-free survival (DFS), while high abundance at the tumor site was associated with poorer OS and DFS. They suggested that Bacteroidales at the extratumoral site might have a protective role in recruiting beneficial T cells and improving the prognosis of colorectal cancer patients, while Bacteroidales at the tumor site could act as pathogens leading to colorectal cancer recurrence. Consistent with these findings, our study also identified Coriobacteriaceae and Porphyromonadaceae as risk factors for colorectal cancer, while Bacteroidales were identified as protective factors. Furthermore, a study conducted by Fortoul et al [56] has identified Hemophilus influenzae as a potential protective factor against colorectal cancer. This observation is intriguingly associated with the up-regulation of NLRP3 inflammasome in response to Hemophilus influenzae infection. The presence of NLRP3 inflammasome may contribute to the maintenance of gut microbiota homeostasis and decrease the risk of colorectal cancer. This intriguing finding suggests a promising avenue for further research.
The gut microbiota performs complex metabolic activities, generating various metabolites that can have both harmful and beneficial effects [57, 58]. These metabolites play a significant role in the interactions between colorectal cancer cells and the gut microbiota. Multiple studies have demonstrated the inhibitory effect of GABA, a metabolite produced by gut microbiota, on the proliferation of colon cancer cells [59]. Furthermore, GABA-producing Lactobacillus plantarum has been shown to induce apoptosis in drug-resistant colorectal cancer cells and inhibit metastasis [60]. In line with these findings, our study also identified GABA as a metabolite associated with a reduced risk of colorectal cancer. However, an animal study revealed that knockdown of the EphB6 gene in mice promoted tumor growth of colorectal cancer cells in a xenograft model by regulating GABA release [61]. Further research is needed to fully understand the role of GABA in colorectal cancer.
In addition to some of the known associations between gut microbiota and digestive tract cancer, we have identified additional causal associations between gut microbiota and these tumors, but no studies have yet reported these associations. This is a novel finding that needs to be explored in further studies. Our study has important implications for pre-cancer screening and intervention. Although much progress has been made in identifying genetic variation in human diseases, most genetic risks remain unexplained. How gut microbiota affects the development and progression of digestive tract cancer still needs to be revealed by further biological studies. Furthermore, while our study primarily sought to explore the causal relationship between gut microbiota and digestive tract cancer, a robust association between gut microbiota and cancers not in the digestive tract is also evident. For instance, Cardeiro et al’s retrospective study identified a statistically significant correlation between enterococcal infection and a decreased occurrence of breast cancer [62]. This research direction holds great promise as well.
Human behavior and the environments in which they live are complex and are influenced by interactions between genes and the environment [63-66]. To eliminate confounding factors in epidemiologic studies, we used MR methods. The SNPs in our study were closely associated with the gut microbiota and were compared with multiple cancer databases. The results of sensitivity analyses showed statistical robustness, and no pleiotropy or heterogeneity was found. However, our study has some limitations. First, the key assumptions of MR have some limitations, and although we tried our best to exclude confounding factors, we cannot fully guarantee the absence of other confounding factors or potential pleiotropic effects. Second, because GWAS pooled data were used, the results may have been affected by different quality control and selection criteria. Third, the analytical principles of MR can only infer potential causal relationships and cannot identify specific biological pathways. Fourth, our study is mainly based on European populations and has limited generalizability to other ethnicities. Finally, due to the lack of data on applicable SNPs, we were unable to explore whether digestive tract cancer leads to alterations in gut microbiota through bidirectional MR studies. Therefore, the possibility of reverse causality needs to be considered with caution in our conclusions and we hope that data on appropriate SNPs will be available in the future for further study interpretation.
Conclusion
Our study conducted a comprehensive evaluation of the association between gut microbiota, gut metabolites, and digestive tract cancer. Our findings suggest that certain gut microbiota and metabolites, when considered as exposure factors, can act as either risk factors or protective factors for digestive tract cancer. These results provide valuable new insights into the mechanisms by which the gut microbiota and gut metabolites influence the development of cancer.
| Supplementary Material | ▴Top |
Table S1. IVs associated with 22 gut microbiota and seven digestive tract cancer (P < 5 × 10-8).
Table S2. IVs associated with 215 gut microbiota and seven digestive tract cancer (P < 1 × 10-5).
Table S3. IVs associated with eight gut metabolites and seven digestive tract cancer (P < 1 × 10-5).
Table S4. SNP number of different gut microbiota and metabolites.
Table S5. Causal effect of MR analysis between 22 gut microbiota and seven digestive tract cancer (P < 5 × 10-8).
Table S6. Causal effects between 22 gut microbiota and seven digestive tract cancer based on IVW method (P < 5 × 10-8).
Table S7. Sensitivity results of MR analysis between 22 gut microbiota and seven digestive tract cancer (P < 5 × 10-8).
Table S8. Causal effect of MR analysis between 215 gut microbiota and seven digestive tract cancer (P < 1 × 10-5).
Table S9. Causal effects between 215 gut microbiota and seven digestive tract cancer based on IVW method (P < 1 × 10-5).
Table S10. Sensitivity results of MR analysis between 215 gut microbiota and seven digestive tract cancer (P < 1 × 10-5).
Table S11. Causal effect of MR analysis between eight gut metabolites and seven digestive tract cancer (P < 1 × 10-5).
Table S12. Causal effects between eight gut metabolites and seven digestive tract cancer based on IVW method (P < 1 × 10-5).
Table S13. Sensitivity results of MR analysis between eight gut metabolites and seven digestive tract cancer (P < 1 × 10-5).
Table S14. MR-PRESSO analysis results of MR analysis between gut microbiota, metabolites and digestive tract cancer.
Fig. 15. Causal effects of gut microbiome and digestive tract cancer based on the IVW (inverse-variance weighted) method (SNPs with P < 1 × 10-5). (A) Heatmap of the P-value. Red color in the heatmap indicates a positive correlation between gut microbiome and digestive tract cancer and blue color indicates a negative correlation. The color depth represents the size of the P-value, with darker colors indicating more significant P-values. (B) Network interactions based on statistically significant odd ratios (ORs). Color and line thickness in the network diagram indicate the OR of gut microbiota and digestive tract cancer. Darker red color and thicker lines indicate higher OR values. (C-I) Forest plot with statistically significant ORs in seven different types of digestive tract cancer. OR > 1 indicates the positive correlation between a particular gut microbiota and a particular digestive tract cancer. OR < 1 indicates the negative correlation between the two. SNP: single nucleotide polymorphism; OR: odds ratio; 95% CI: 95% confidence interval; NSNP: number of SNP.
Acknowledgments
The authors thank all investigators and participants from the MiBioGen, TwinsUK Registry and FinnGen research contributing to GWAS of gut microbiota, gut metabolites, and digestive tract cancer.
Financial Disclosure
This project was funded by Administration of Traditional Chinese Medicine of Guangdong Province (Grant No. 20231078).
Conflict of Interest
The authors declare that they have no competing interests.
Informed Consent
Not applicable.
Author Contributions
Jin Sheng Huang and Xu Jia Li were involved in the conception and design; Jin Sheng Huang, Xu Jia Li, Meng Ge Gao, Ling Li Huang, Xu Xian Chen, and Yu Ming Rong were involved in analysis and interpretation of the data; Jin Sheng Huang and Ling Li Huang were involved in the drafting of the paper or revising it critically for intellectual content. All authors agree to be accountable for all aspects of the work.
Data Availability
The summary statistics of exposures were available on the MiBioGen and TwinsUK Registry; the summary statistics of outcome were available on the FinnGen research (https://www.finngen.fi/fi).
| References | ▴Top |
- Korpela K, de Vos WM. Early life colonization of the human gut: microbes matter everywhere. Curr Opin Microbiol. 2018;44:70-78.
doi pubmed - Korpela K, Helve O, Kolho KL, Saisto T, Skogberg K, Dikareva E, Stefanovic V, et al. Maternal fecal microbiota transplantation in cesarean-born infants rapidly restores normal gut microbial development: a proof-of-concept study. Cell. 2020;183(2):324-334.e325.
doi pubmed - Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, Brady A, et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. 2017;550(7674):61-66.
doi pubmed pmc - Qin Y, Havulinna AS, Liu Y, Jousilahti P, Ritchie SC, Tokolyi A, Sanders JG, et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat Genet. 2022;54(2):134-142.
doi pubmed pmc - de Vos WM, Tilg H, Van Hul M, Cani PD. Gut microbiome and health: mechanistic insights. Gut. 2022;71(5):1020-1032.
doi pubmed pmc - Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, Watanabe H, et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med. 2019;25(6):968-976.
doi pubmed - Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101(1):5-22.
doi pubmed pmc - Sekula P, Del Greco MF, Pattaro C, Kottgen A. Mendelian Randomization as an Approach to Assess Causality Using Observational Data. J Am Soc Nephrol. 2016;27(11):3253-3265.
doi pubmed pmc - Smith GD, Ebrahim S. 'Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1-22.
doi pubmed - Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89-98.
doi pubmed pmc - Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362:k601.
doi pubmed pmc - Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, Le Roy CI, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156-165.
doi pubmed pmc - Moayyeri A, Hammond CJ, Valdes AM, Spector TD. Cohort Profile: TwinsUK and healthy ageing twin study. Int J Epidemiol. 2013;42(1):76-85.
doi pubmed pmc - Andersson C, Johnson AD, Benjamin EJ, Levy D, Vasan RS. 70-year legacy of the Framingham Heart Study. Nat Rev Cardiol. 2019;16(11):687-698.
doi pubmed - Rhee EP, Ho JE, Chen MH, Shen D, Cheng S, Larson MG, Ghorbani A, et al. A genome-wide association study of the human metabolome in a community-based cohort. Cell Metab. 2013;18(1):130-143.
doi pubmed pmc - Peltonen L, Jalanko A, Varilo T. Molecular genetics of the Finnish disease heritage. Hum Mol Genet. 1999;8(10):1913-1923.
doi pubmed - Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658-665.
doi pubmed pmc - Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985-1998.
doi pubmed pmc - Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512-525.
doi pubmed pmc - Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304-314.
doi pubmed pmc - Verbanck M, Chen CY, Neale B, Do R. Publisher Correction: Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(8):1196.
doi pubmed - Gupta V, Walia GK, Sachdeva MP. 'Mendelian randomization': an approach for exploring causal relations in epidemiology. Public Health. 2017;145:113-119.
doi pubmed - Burgess S, Thompson SG. Bias in causal estimates from Mendelian randomization studies with weak instruments. Stat Med. 2011;30(11):1312-1323.
doi pubmed - Bowden J, Del Greco MF, Minelli C, Zhao Q, Lawlor DA, Sheehan NA, Thompson J, et al. Improving the accuracy of two-sample summary-data Mendelian randomization: moving beyond the NOME assumption. Int J Epidemiol. 2019;48(3):728-742.
doi pubmed pmc - Nan H, Morikawa T, Suuriniemi M, Imamura Y, Werner L, Kuchiba A, Yamauchi M, et al. Aspirin use, 8q24 single nucleotide polymorphism rs6983267, and colorectal cancer according to CTNNB1 alterations. J Natl Cancer Inst. 2013;105(24):1852-1861.
doi pubmed pmc - Nan H, Hutter CM, Lin Y, Jacobs EJ, Ulrich CM, White E, Baron JA, et al. Association of aspirin and NSAID use with risk of colorectal cancer according to genetic variants. JAMA. 2015;313(11):1133-1142.
doi pubmed pmc - Khalili H, Gong J, Brenner H, Austin TR, Hutter CM, Baba Y, Baron JA, et al. Identification of a common variant with potential pleiotropic effect on risk of inflammatory bowel disease and colorectal cancer. Carcinogenesis. 2015;36(9):999-1007.
doi pubmed pmc - Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148(6):1258-1270.
doi pubmed pmc - Huang J, Li X, Hong J, Huang L, Jiang Q, Guo S, Rong Y, et al. Inflammatory bowel disease increases the risk of hepatobiliary pancreatic cancer: A two-sample Mendelian randomization analysis of European and East Asian populations. Cancer Med. 2023;12(12):13599-13609.
doi pubmed pmc - Mima K, Kosumi K, Baba Y, Hamada T, Baba H, Ogino S. The microbiome, genetics, and gastrointestinal neoplasms: the evolving field of molecular pathological epidemiology to analyze the tumor-immune-microbiome interaction. Hum Genet. 2021;140(5):725-746.
doi pubmed pmc - Seo SK, Kwon B. Immune regulation through tryptophan metabolism. Exp Mol Med. 2023;55(7):1371-1379.
doi pubmed pmc - Huang C, Mei S, Zhang X, Tian X. Inflammatory milieu related to dysbiotic gut microbiota promotes tumorigenesis of hepatocellular carcinoma. J Clin Gastroenterol. 2023;57(8):782-788.
doi pubmed - Tsuruya A, Kuwahara A, Saito Y, Yamaguchi H, Tenma N, Inai M, Takahashi S, et al. Major anaerobic bacteria responsible for the production of carcinogenic acetaldehyde from ethanol in the colon and rectum. Alcohol Alcohol. 2016;51(4):395-401.
doi pubmed - Wang L, Tu YX, Chen L, Zhang Y, Pan XL, Yang SQ, Zhang SJ, et al. Male-biased gut microbiome and metabolites aggravate colorectal cancer development. Adv Sci (Weinh). 2023;10(25):e2206238.
doi pubmed pmc - Cheung MK, Yue GGL, Lauw S, Li CSY, Yung MY, Ng SC, Yip HC, et al. Alterations in gut microbiota of esophageal squamous cell carcinoma patients. J Gastroenterol Hepatol. 2022;37(10):1919-1927.
doi pubmed - Eun CS, Kim BK, Han DS, Kim SY, Kim KM, Choi BY, Song KS, et al. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter. 2014;19(6):407-416.
doi pubmed - Zi M, Zhang Y, Hu C, Zhang S, Chen J, Yuan L, Cheng X. A literature review on the potential clinical implications of streptococci in gastric cancer. Front Microbiol. 2022;13:1010465.
doi pubmed pmc - Ayanaba A, Alexander M. Microbial formation of nitrosamines in vitro. Appl Microbiol. 1973;25(6):862-868.
doi pubmed pmc - Coker OO, Dai Z, Nie Y, Zhao G, Cao L, Nakatsu G, Wu WK, et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut. 2018;67(6):1024-1032.
doi pubmed pmc - Abate M, Vos E, Gonen M, Janjigian YY, Schattner M, Laszkowska M, Tang L, et al. A novel microbiome signature in gastric cancer: a two independent cohort retrospective analysis. Ann Surg. 2022;276(4):605-615.
doi pubmed pmc - Tseng CH, Lin JT, Ho HJ, Lai ZL, Wang CB, Tang SL, Wu CY. Gastric microbiota and predicted gene functions are altered after subtotal gastrectomy in patients with gastric cancer. Sci Rep. 2016;6:20701.
doi pubmed pmc - Sasaki H, Ishizuka T, Muto M, Nezu M, Nakanishi Y, Inagaki Y, Watanabe H, et al. Presence of Streptococcus anginosus DNA in esophageal cancer, dysplasia of esophagus, and gastric cancer. Cancer Res. 1998;58(14):2991-2995.
pubmed - Sasaki H, Igaki H, Ishizuka T, Kogoma Y, Sugimura T, Terada M. Presence of Streptococcus DNA sequence in surgical specimens of gastric cancer. Jpn J Cancer Res. 1995;86(9):791-794.
doi pubmed pmc - Zhang X, Li C, Cao W, Zhang Z. Alterations of gastric microbiota in gastric cancer and precancerous stages. Front Cell Infect Microbiol. 2021;11:559148.
doi pubmed pmc - Jia X, Lu S, Zeng Z, Liu Q, Dong Z, Chen Y, Zhu Z, et al. Characterization of gut microbiota, bile acid metabolism, and cytokines in intrahepatic cholangiocarcinoma. Hepatology. 2020;71(3):893-906.
doi pubmed - Ma Y, Nenkov M, Chen Y, Press AT, Kaemmerer E, Gassler N. Fatty acid metabolism and acyl-CoA synthetases in the liver-gut axis. World J Hepatol. 2021;13(11):1512-1533.
doi pubmed pmc - Jiang N, Song X, Peng YM, Wang WN, Song Z. Association of disease condition with changes in intestinal flora, and plasma endotoxin and vascular endothelial growth factor levels in patients with liver cancer. Eur Rev Med Pharmacol Sci. 2020;24(7):3605-3613.
doi pubmed - Yang J, He Q, Lu F, Chen K, Ni Z, Wang H, Zhou C, et al. A distinct microbiota signature precedes the clinical diagnosis of hepatocellular carcinoma. Gut Microbes. 2023;15(1):2201159.
doi pubmed pmc - Zhong H, Liu S, Zhu J, Wu L. Associations between genetically predicted levels of blood metabolites and pancreatic cancer risk. Int J Cancer. 2023;153(1):103-110.
doi pubmed - Yang J, Ma Y, Tan Q, Zhou B, Yu D, Jin M, Zhang T, et al. Gut Streptococcus is a microbial marker for the occurrence and liver metastasis of pancreatic cancer. Front Microbiol. 2023;14:1184869.
doi pubmed pmc - Ogrendik M. The association between oral anaerobic bacteria and pancreatic cancer. World J Oncol. 2023;14(3):174-177.
doi pubmed pmc - Qu R, Zhang Y, Ma Y, Zhou X, Sun L, Jiang C, Zhang Z, et al. Role of the gut microbiota and its metabolites in tumorigenesis or development of colorectal cancer. Adv Sci (Weinh). 2023;10(23):e2205563.
doi pubmed pmc - Yu I, Wu R, Tokumaru Y, Terracina KP, Takabe K. The role of the microbiome on the pathogenesis and treatment of colorectal cancer. Cancers (Basel). 2022;14(22):5685.
doi pubmed pmc - Borges-Canha M, Portela-Cidade JP, Dinis-Ribeiro M, Leite-Moreira AF, Pimentel-Nunes P. Role of colonic microbiota in colorectal carcinogenesis: a systematic review. Rev Esp Enferm Dig. 2015;107(11):659-671.
doi pubmed - Huo RX, Wang YJ, Hou SB, Wang W, Zhang CZ, Wan XH. Gut mucosal microbiota profiles linked to colorectal cancer recurrence. World J Gastroenterol. 2022;28(18):1946-1964.
doi pubmed pmc - Fortoul MC, Kim E, Ardeljan AD, Frankel L, Takabe K, Rashid OM. The role of hemophilus influenzae infection and its relationship with colorectal cancer. World J Oncol. 2023;14(3):188-194.
doi pubmed pmc - Tang WH, Kitai T, Hazen SL. Gut microbiota in cardiovascular health and disease. Circ Res. 2017;120(7):1183-1196.
doi pubmed pmc - Zhang B, Li J, Fu J, Shao L, Yang L, Shi J. Interaction between mucus layer and gut microbiota in non-alcoholic fatty liver disease: Soil and seeds. Chin Med J (Engl). 2023;136(12):1390-1400.
doi pubmed pmc - Song L, Du A, Xiong Y, Jiang J, Zhang Y, Tian Z, Yan H. gamma-Aminobutyric acid inhibits the proliferation and increases oxaliplatin sensitivity in human colon cancer cells. Tumour Biol. 2016;37(11):14885-14894.
doi pubmed - An J, Seok H, Ha EM. GABA-producing Lactobacillus plantarum inhibits metastatic properties and induces apoptosis of 5-FU-resistant colorectal cancer cells via GABA(B) receptor signaling. J Microbiol. 2021;59(2):202-216.
doi pubmed - Yu H, Qin XK, Yin KW, Li ZM, Ni ED, Yang JM, Liu XH, et al. EphB6 deficiency in intestinal neurons promotes tumor growth in colorectal cancer by neurotransmitter GABA signaling. Carcinogenesis. 2023.
doi pubmed - Cardeiro M, Ardeljan AD, Frankel L, Kim E, Takabe K, Rashid OM. Incidence of breast cancer and enterococcus infection: a retrospective analysis. World J Oncol. 2023;14(1):32-39.
doi pubmed pmc - Meisel SF, Beeken RJ, van Jaarsveld CH, Wardle J. Genetic susceptibility testing and readiness to control weight: Results from a randomized controlled trial. Obesity (Silver Spring). 2015;23(2):305-312.
doi pubmed pmc - Tucker-Drob EM, Bates TC. Large cross-national differences in gene x socioeconomic status interaction on intelligence. Psychol Sci. 2016;27(2):138-149.
doi pubmed pmc - Agusti A, Melen E, DeMeo DL, Breyer-Kohansal R, Faner R. Pathogenesis of chronic obstructive pulmonary disease: understanding the contributions of gene-environment interactions across the lifespan. Lancet Respir Med. 2022;10(5):512-524.
doi pubmed - Meisel SF, Walker C, Wardle J. Psychological responses to genetic testing for weight gain: a vignette study. Obesity (Silver Spring). 2012;20(3):540-546.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.