

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website http://www.wjon.org |
Original Article
Volume 9, Number 4, August 2018, pages 97-103
Age-Related Chromosomal Aberrations in Patients with Diffuse Large B-Cell Lymphoma: An In Silico Approach
Eric J. Vicka, Noah Richardsonb, Kruti Patelc, Glenda M. Delgado Ramosc, Alaa Altahanc, Taylor Allowayc, Michael G. Martind, e
aDepartment of Internal Medicine, University of Cincinnati, Cincinnati OH, USA
bCollege of Medicine, The University of Tennessee Health Science Center, Memphis, TN, USA
cDepartment of Internal Medicine, The University of Tennessee Health Science Center, Memphis, TN, USA
dDepartment of Hematology and Oncology, The West Cancer Center, Memphis, TN, USA
eCorresponding Author: Michael Martin, Department of Hematology and Oncology, The West Cancer Center, Memphis, TN, USA
Manuscript submitted July 17, 2018, accepted August 27, 2018
Short title: Age Aberrations in DLBCL
doi: https://doi.org/10.14740/wjon1136w
| Abstract | ▴Top |
Background: In diffuse large B-cell lymphoma (DLBCL), chromosomal aberrations are known to increase with advancing age. Our study aims to determine if there are other genetic aberrations associated with DLBCL based on age.
Methods: Using the Mitelman Database of Genetic Aberrations, we were able to find 749 cases of DLBCL with genomic aberrations with a median age of 62 years. Patients with DLBCL chromosomal aberration analysis results were divided into four groups based on age (0 - 30, 31 - 50, 51 - 70, > 71 years) and examined by chi-square analysis and Mantel-Cox for survival analysis.
Results: Ten aberrations were found to be significant with a particular age range: t(2;3), trisomy 19p13, trisomy 18q21, trisomy 3, trisomy 7, trisomy 14, trisomy 16, trisomy 18, monosomy 3 and monosomy 11, and survival ranged from 7 to 25 months.
Conclusion: This suggests that patients with DLBCL are likely to accumulate specific translocations depending on their age at the onset of DLBCL.
Keywords: Cancer and aging; Genetic aberrations; Diffuse large B-cell Lymphoma
| Introduction | ▴Top |
Chromosomal instability and rearrangements may occur at any age, but many aberrations are known to confer a selective advantage in diffuse large B-cell lymphoma (DLBCL). Aberrations which amplify the effect of proto-oncogenes (t(3;14), t(8;14), t(14;18), t(15;17), trisomy 3q27) and those which reduce or prevent the effects of tumor suppressor genes (deletions of chromosome 17, 16p11) have profound effects on cellular behavior and are the inciting events that lead to cancer. While many chromosome aberrations have a straight forward effect (c-MYC amplification in t(8;14)), others have a role that has yet to be determined clearly such as monosomy 19p13 or a seemingly paradoxical role seen in the deletion or addition of chromosome 3. While in some cases aberrations may be benign, it is imperative that we understand more about how these aberrations at the chromosomal, genetic and epigenetic level affect survival and susceptibility in the era of personalized medicine.
DLBCL is the most common subtype of non-Hodgkin lymphoma in adults and includes a heterogeneous group of subtypes. While DLBCL may arise de novo, it may also result from the transformation of more indolent lymphomas such as chronic lymphocytic leukemia, lymphocyte predominant Hodgkin lymphoma, marginal zone lymphoma and follicular lymphoma [1, 2]. The characteristic marking the change from any of these more indolent lymphomas into DLBCL is the loss of histological architecture of the former to the diffuse unregulated architecture of DLBCL along with a clonal relationship. DLBCL tends to develop later in life, with a median age of onset around 70, though younger patients tend to have better outcomes, with age ≥ 70 associated with decreased overall survival [3]. However, 2-year progression-free survival (PFS) has improved dramatically in the post rituximab era (78%), and may improve further with the use of highly specified therapies [3, 4]. As molecular diagnosis advances, it is essential to learn more about how the genetic landscape of DLBCL can influence potential therapeutic options, and their presentation according to age.
Younger patients with DLBCL have a better overall prognosis, which is likely due to a variety of factors including fewer comorbidities, decreased overall genetic damage and aberrations with a superior overall prognosis [5, 6]. This is similar to related diseases such as acute myeloid leukemia (AML) [7] and acute lymphoblastic leukemia (ALL), with studies suggesting that socioeconomic factors do not as much of a role as the intrinsic makeup of the disease [8]. In such a case, individual mutations and aberrations may be a marker for a particular type of malignancy. For example, anaplastic lymphoma kinase (ALK)-positive anaplastic large-cell lymphoma (ALCL) generally affects a younger population with a median age of 17 years and the expression of the ALK fusion protein is a positive prognostic indicator [9]. ALK positivity is general associated with a younger population, while others are associated with tumor suppressor genes like VHL on chromosome 3 or ATM on chromosome 11 [10, 11].
Certain genetic aberrations in DLBCL have become essential to diagnosis and treatment of DLBCL. Double hit and triple hit lymphomas involving translocations or aberrations of c-MYC, BCL2 and BCL6 are known to carry a poor prognosis [12]. c-MYC is located on chromosome 8q24 seen in 3-16% of DLBCL, with double hit lymphomas occurring 0-12% of the time [13]. A study conducted by Savage et al [12] demonstrated a poor prognosis in DLBCL with t(8;14) MYC rearrangements, with an average age of 69 years. Furthermore, the copy number of MYC in these cases was also found to be independently associated with a poor prognosis [14]. DLBCL with t(14;18) translocations involving BCL2 is found almost exclusively in patients older than 18 years of age [15], and advancing age in general is associated with the development of micronuclei, aneuploidy and increasing chromosomal aberration [16]. In a large-scale chromosomal study of 60 cases of myeloid malignancies with t(2;3), it was found that patients commonly present with concomitant abnormalities that were clinically relevant with poorer prognosis, predominantly monosomy 7, deletion of 5q and t(9;22) [17]. Very old age (> 81 years) is an independent risk factor for poor overall survival in DLBCL.
Our study aims to determine if there are other specific genetic aberrations associated with certain age groups. Development of specific aberrations and their relevance to clinical outcomes have been discussed at length; however, there is sparse information on the development of aberrations as they relate to young, middle aged or elderly patients. The age of diagnosis may play an important role in offering both prognostic value and molecular characteristics which help to identify patients who are at risk.
| Patients and Methods | ▴Top |
Data acquisition and search terms
The National Cancer Institute’s Mitelman Database of Chromosome Aberrations and Gene Fusions was used to identify patients with DLBCL. On the date of inquiry (March 1, 2017), this represented 1,006 Pts with DLBCL. Publications before 1990 were excluded for data consistency, leaving a total of 749 patients. One hundred twenty-eight different parameters of inquiry (Supplementary Table 1) were used to identify specific chromosomal aberrations and these aberrations were divided into four age groups (0 - 30, 31 - 50, 51 - 70, > 71 years). Significance was determined by chi-square analysis, where P < 0.05 was determined to be significant. Aberrations concluded to be non-significant due low incidence were excluded from the survival analysis but are mentioned in the text. Calculations of chromosome complexity utilized each comma as a delimiter of the aberration. Chromosome number was determined by either the number provided or the average number of those provided.
 Click to view | Table 1. Overview of Patient Characteristics |
Survival analysis
Survival analysis was conducted based on available data from publications. Survival was calculated from the date of diagnosis to the date of death or follow-up. Data that did mention date of death or follow-up was omitted. Mantel-Cox log-rank and Gehan-Breslow-Wilcoxon tests were used for survival analysis and significance between curves. All P values were two-sided and an alpha value of less than 0.05 was used for significance. Statistical analysis was conducted using Graphpad Prism 7.
| Results | ▴Top |
From the overall query of 128 different terms, a total of 10 aberrations were identified which showed a significant association with a particular age range (9.4%). The translocation t(2;3) was associated with a younger age range. Two were chromosomal portions (19p13 and 18q21) which were both associated with older age ranges. The vast majority of significant data points involved whole chromosome duplications (3, 7, 14, 16 and 18) and deletions (3, 11) (Table 1). Altogether, this demonstrates that while the mechanism of aberrations may vary, certain aberrations are more likely to occur in specific age groups.
Younger patients were also more likely to have a translocation associated with their DLBCL. Translocations of chromosome 2 were associated with younger patients as 60% of the patients with t(2;3) translocation were under 50 (Fig. 1). The t(1;12) translocation showed a trend toward younger patients but lacked a large enough sample size to be considered significant. There were examples of increasing incidence with age as well. Trisomy 19p13 and trisomy 18 were conspicuous examples, with increasing incidence in older patients. In addition, there were two aberrations (t(7;16) and trisomy 2p23), which showed high proportions of late middle age and elderly patients but lacked an adequate sample size for significance.
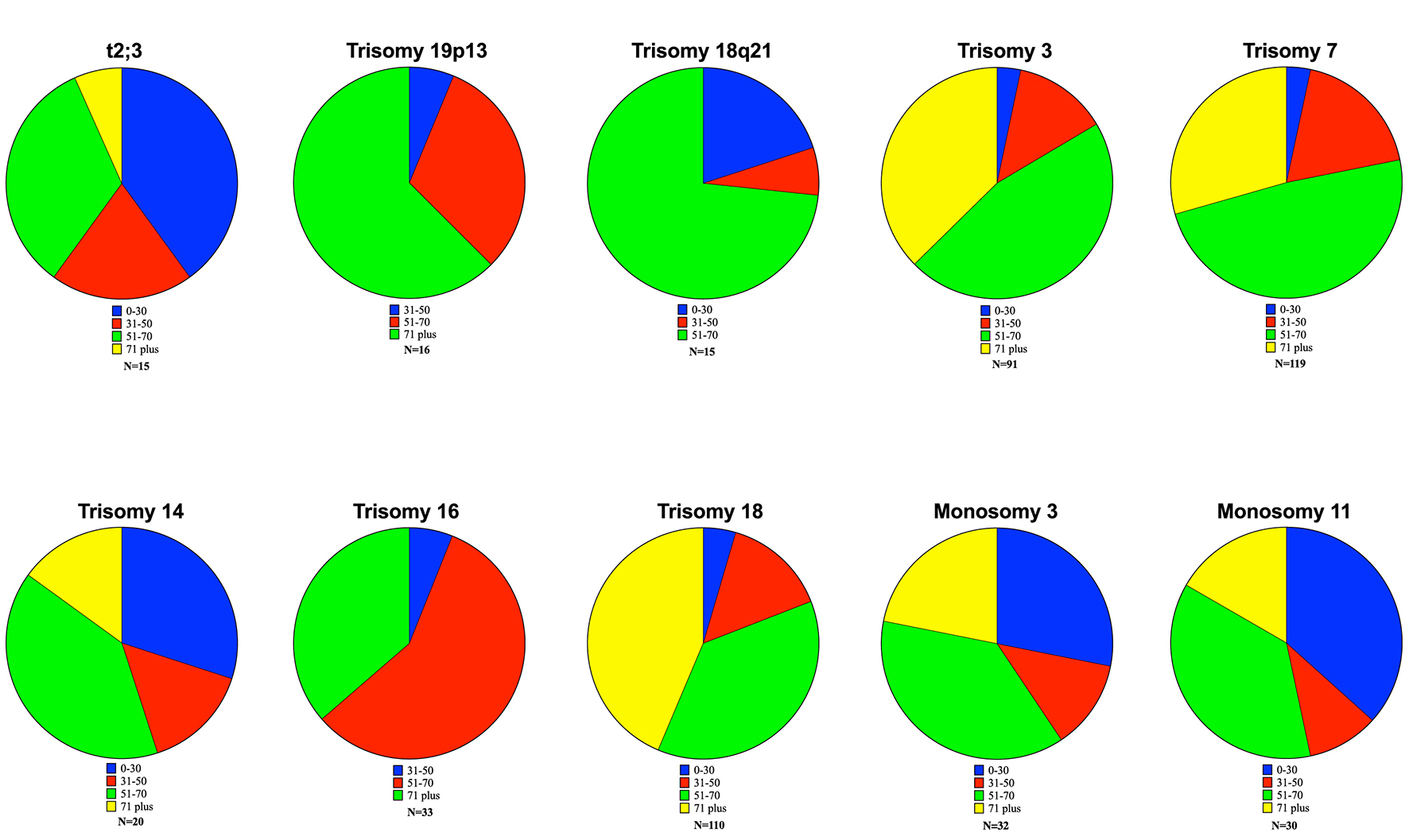 Click for large image | Figure 1. Age-based proportions of significant aberrations. Ten significant data points were obtained. The t(2;3) translocation showed a significant increase in the proportion of patients less than 30. Similar findings were observed in trisomy 14, monosomy 3 and monosomy 11. Older patients (> 71 years) showed increased incidence of the additions of 19p13, 18q21, trisomy 3, trisomy 16 and trisomy 18. |
Our results demonstrated a large degree of variability within our DLBCL population, with a mean chromosome number of 59.8, ranging from 36 to 109.5. The total number of aberrations was also variant, with a mean of 8.6, ranging from 1 to 51 per case. Results for mean chromosomal number were also varied from 46.5 (trisomy 18q21) to 73.5 (monosomy 11). The average number of aberrations within any sample was also variant. The mean number of aberrations was 8.6 within the entire sample population. The aberrations which we found to be significant tended to have increased chromosomal instability compared to the sample population as a whole and ranged from 8.2 (trisomy 3) to 18.8 (monosomy 11).
Our study results were from a range of studies published between 1990 and 2016 and the median overall date of publication was 2000. Survival was determined from these publications when provided (Fig. 2). The median overall survival was 14 months for the entire population, and differences in survival among significant aberrations were not significant (log-rank test, P = 0.7512, Gahen-Breslow-Wilcoxon Test, P = 0.8759). Aberrations with the shortest median survival were trisomy 19p13 (7 months) and trisomy 16 (9 months). The longest survival occurred in trisomy 14 (25 months), monosomy 11 (21 months), trisomy 3 and trisomy 7 (19 months).
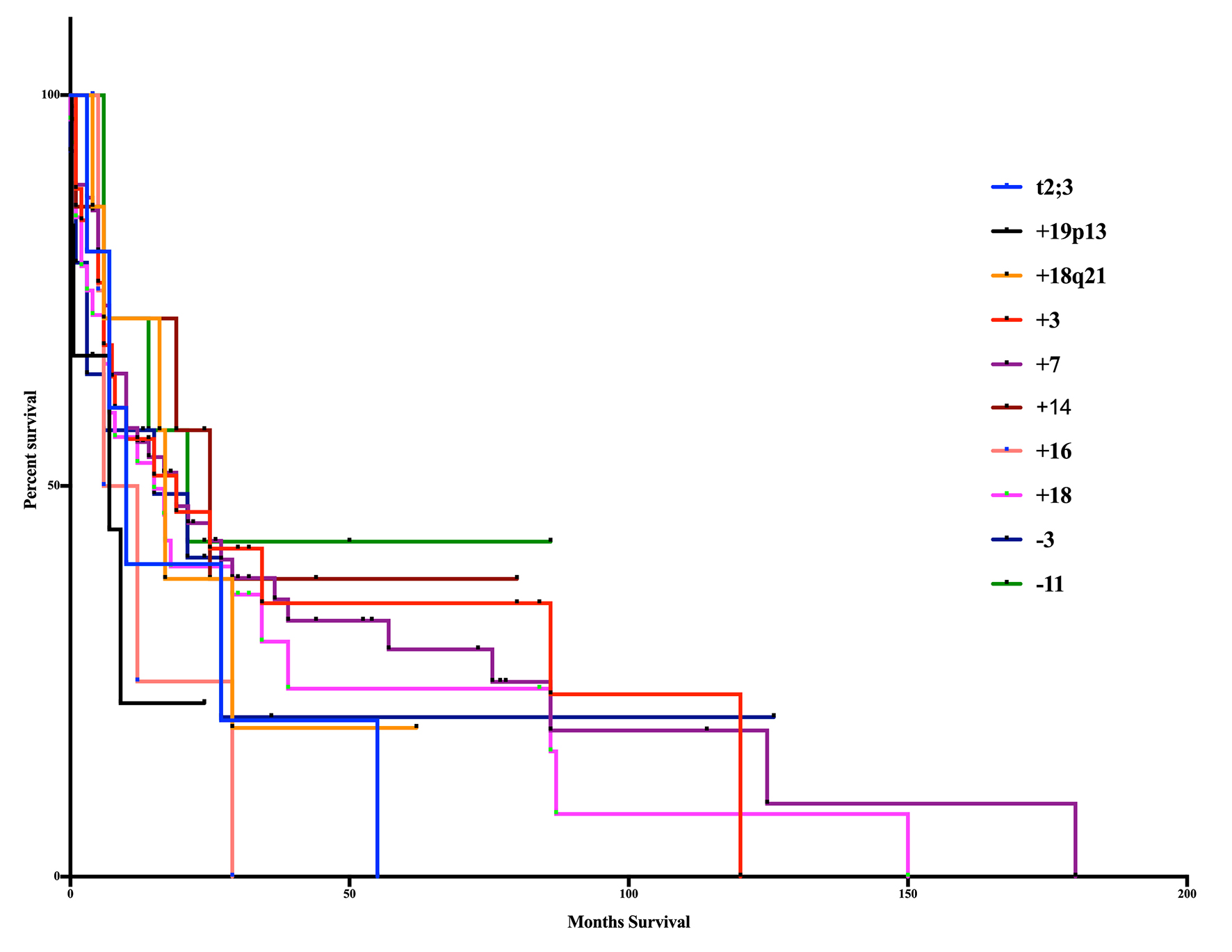 Click for large image | Figure 2. Kaplan-Meier curve of age-based aberrations. The 10 significant aberrations did not show any significant differences from one another (log-rank test, P = 0.7512, Gahen-Breslow-Wilcoxon test, P = 0.8759). The poorest overall survival occurred in patients with the addition of 19p13 (7m) and trisomy 16 (9m). Superior survival occurred in those patients in trisomy 14 (25m), monosomy 11 (21m), trisomy 3 (19m) and trisomy 7 (19m). |
| Discussion | ▴Top |
Our study shows that age is a factor when considering the chromosomal aberrations associated with DLBCL. Ten significant age-related aberrations were found in DLBCL, some of which have been previously demonstrated, while others are novel (19p13, 18q21). The t(2;5) translocation, which transposes nucleophosmin to the ALK gene, has rarely been associated with younger patients in the ALK+ variant of DLBCL [18, 19]. However, a comprehensive systematic review of 108 ALK+ DLBCL only found four patients with NPM-ALK t(2,5). NPM-ALK is the most common chimeric protein in ALK+ ALCL, but it is rare in ALK+ DLBCL, which the majority show the t(2;17) CLTC-ALK fusion gene [19, 20]. Our t(2;5) translocation results likely reflect misdiagnosis of ALK+ ALCL and therefore, were not included as significant findings. Similarly, we found many instances of the t(8;14) translocation which involves many iterations of the c-MYC gene with the immunoglobulin heavy chain locus [20]. Our analysis demonstrated a proportionately significant increase in patients under the age of 30 years, and with a decrease in incidence in patients over 71 years, as well as a median survival of 8 months for this population, but again many of these are likely misdiagnosed or miscoded Burkitt lymphoma given the population age.
A population we found to be associated with younger patients was the t(2;3) translocation which often combines the immunoglobulin κ light chain gene and the BCL6 protein (median age: 54.5 years). In our study, there were also several cases of the EVI fusion gene fused with ETV6, which is more common in cases of myeloid malignancies [21]. While there have been many case studies of the t(2;3) translocation in DLBCL, few have looked at age as an isolated factor. BCL6 aberrations of some kind are known to occur in roughly 20% of DLBCL [22], but the incidence of the t(2;3) translocations remains fairly rare (2.9%) [23]. Survival differences, especially between the pre-and post-rituximab era, are substantial. The median survival of our t(2;3) translocation population was 10 months, which we believe to be a novel estimate in DLBCL (Fig. 2).
The 19p13 locus has been found to be associated with multiple cancer types [24]. In all cases, including DLBCL, the exact gene or genes which cause it to be a frequent target are unknown. Several targets have been suggested including TCF3 [25], TNFSF7, TNFSF9 [26], as well as several mircoRNAs located in the region [27]. The most direct comparison conducted by Kim et al [28], found 19p13 to be a poor prognostic indicator but did not elaborate on the age of the population (16). They found that 19p13 aberrations in general were associated with a low overall survival, with a 95% confidence interval of between 1.50 and 4.76 months. Our study, with an identical number of patients, found a gain of 19p13 to increase with age, with the majority of patients over 71 years and zero patients under 30 years. We also found it to be a poor prognostic indicator with a mean overall survival of 7 months, which is likely associated with increasing age of the patients involved.
Chromosome 18q21 includes the paracaspase MALT-1, which in its activated form leads to the activation of BCL-10 and NF-κB. This in turn leads to an upregulation of BCL-2 in the absence of further genetic modification, including the t(14;18) translocation [29, 30]. In previous studies, gain of 18q21 functioned as an independent negative predictive factor [30], while in other studies, it was found to be non-significant in contribution [28, 31]. A separate study showed that relapses were increased in patients who gained 18q21 [32]. Our study shows that the majority of patients were between the ages of 51 and 70 years (73.3%) and median survival was actually improved compared to our total population, though not significantly (17 months). MALT1 along with its signaling partners has recently become a target of small molecular inhibitors which are showing potent activity in preclinical screening [33].
Chromosome instability is well known in oncology, caused by increasing cellular dysregulation and natural selection of new clones [32]. Whole chromosome aberrations are a well-known but poorly defined mechanism to amplify specific genes such as BCL2 by increasing the number of chromosome 18 but determining with certainty which genes are involved in highly speculative. Trisomy 3 [34], 14 [35], 16 [36] and 18 [37] are often found in hematological malignancies, and their incidence has been associated primarily with the proto-oncogenes found on those chromosomes. We found both a gain and loss of chromosome 3 to be significant, though monosomy 3 was associated with an increase in patients under 30 years, while trisomy 3 occurred primarily in patients over 50 years. Survival was only lessened in the case of trisomy 16 (9 months), while all others were above the median of the population. Trisomy 14, monosomy 3 and monosomy 11 were all enriched in patients under of the age of 30 years. Chromosomes 3 and 11 are the locations of the VHL and ATM tumor suppressor genes, which could shed some light on their respective deletions. ATM is produced in response to DNA damage, which leads to downstream activation of DNA repair enzymes and p53. It is known that mutations and variants of ATM may lead to malignancy, so by selecting for clones with a deletion of chromosome 11, it is possible for this to be a reason for increase in genetic aberrations seen in that population [38].
Though we found several novel associations, our study is not without limitations. Firstly, our observations were recorded retrospectively from many different sources. The numbers of patients in some groups both for survival and chromosomal analyses had a low overall number of patients. For three groups of chromosomal analysis, 16 or fewer patients were identified, and fewer were identified with corresponding survival information (t2;3 translocation, trisomy 19p13 and trisomy 18q21). In these cases, further study is needed to understand the utility of these findings.
In addition, these sources have different prerogatives for their data, so many cases did not have survival information (Supplementary Table 1). In those that did, we only recorded the survival information where the survival could be determined from the date of diagnosis to the date of death. Our observations also only considered the single aberrations that occurred and not the complete karyotype of each patient, which obviously complicates drawing conclusions regarding prognosis. Finally, while limiting the starting year of our study to 1990, it bridges the pre- and post-rituximab era and some data points may be biased more toward one treatment paradigm. Our previous data [39] had shown some significant points which, by restricting the data to 1990, were no longer significant. This is likely due to differences in diagnostic and clinical assessment before 1990, and many similar issues likely apply to our current data in a less profound way while preserving a fairly large patient dataset. Despite these limitations, we believe that our data provides novel insight into the age associations of particular aberrations in DLBCL.
The association of chromosomal aberrations with DLBCL has been long established; however, therapies are now being employed to target and stratify risk of specific therapies [40]. Moving forward, it is essential to understand how these abnormalities contribute to diagnostic and prognostic features both in DLBCL and in cancer in general. Further investigation is necessary to look for the associated genetic changes at a smaller scale as well as the epigenetic changes that may be influencing these aberrations.
Author Contributions
EV, NR and MM designed and wrote the manuscript. EV, NR, KP and MM were all involved in the conceptual design and analysis planning of the project. EV, NR, KP, TA, GDR, AA and MM were involved in acquisition of the data. EV performed the statistical analysis of the data. EV, NR and MM drafted the manuscript. EV, NR, KP and MM were all involved in critical revision of the manuscript. MM supervised all aspects of the study.
Disclosure
The authors have nothing to disclose.
| References | ▴Top |
- Martelli M, Ferreri AJ, Agostinelli C, Di Rocco A, Pfreundschuh M, Pileri SA. Diffuse large B-cell lymphoma. Crit Rev Oncol Hematol. 2013;87(2):146-171.
doi pubmed - Testoni M, Zucca E, Young KH, Bertoni F. Genetic lesions in diffuse large B-cell lymphomas. Ann Oncol. 2015;26(6):1069-1080.
doi pubmed - Saygin C, Jia X, Hill B, Dean R, Pohlman B, Smith MR, Jagadeesh D. Impact of comorbidities on outcomes of elderly patients with diffuse large B-cell lymphoma. Am J Hematol. 2017;92(10):989-996.
doi pubmed - Leonard JP, Kolibaba KS, Reeves JA, Tulpule A, Flinn IW, Kolevska T, Robles R, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-Cell-Like diffuse large B-Cell lymphoma. J Clin Oncol. 2017;35(31):3538-3546.
doi pubmed - Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, et al. Axicabtagene ciloleucel CAR T-Cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531-2544.
doi pubmed - Sandlund JT, Martin MG. Non-Hodgkin lymphoma across the pediatric and adolescent and young adult age spectrum. Hematology Am Soc Hematol Educ Program. 2016;2016(1):589-597.
doi pubmed - Nasir SS, Giri S, Nunnery S, Martin MG. Outcome of adolescents and young adults compared with pediatric patients with acute myeloid and promyelocytic leukemia. Clin Lymphoma Myeloma Leuk. 2017;17(2):126-132 e121.
- Fintel AE, Jamy O, Martin MG. Influence of insurance and marital status on outcomes of adolescents and young adults with acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk. 2015;15(6):364-367.
doi pubmed - Nomura M, Narita Y, Miyakita Y, Ohno M, Fukushima S, Maruyama T, Muragaki Y, et al. Clinical presentation of anaplastic large-cell lymphoma in the central nervous system. Mol Clin Oncol. 2013;1(4):655-660.
doi pubmed - Vicha A, Holzerova M, Krepelova A, Musil Z, Prochazka P, Sumerauer D, Kodet R, et al. Molecular cytogenetic characterization in four pediatric pheochromocytomas and paragangliomas. Pathol Oncol Res. 2011;17(4):801-808.
doi pubmed - Luskin M, Wertheim G, Morrissette J, Daber R, Biegel J, Wilmoth D, Kersun L, et al. CLL/SLL diagnosed in an adolescent. Pediatr Blood Cancer. 2014;61(6):1107-1110.
doi pubmed - Savage KJ, Johnson NA, Ben-Neriah S, Connors JM, Sehn LH, Farinha P, Horsman DE, et al. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood. 2009;114(17):3533-3537.
doi pubmed - Aukema SM, Siebert R, Schuuring E, van Imhoff GW, Kluin-Nelemans HC, Boerma EJ, Kluin PM. Double-hit B-cell lymphomas. Blood. 2011;117(8):2319-2331.
doi pubmed - Quesada AE, Medeiros LJ, Desai PA, Lin P, Westin JR, Hawsawi HM, Wei P, et al. Increased MYC copy number is an independent prognostic factor in patients with diffuse large B-cell lymphoma. Mod Pathol. 2017;30(12):1688-1697.
doi pubmed - Oschlies I, Klapper W, Zimmermann M, Krams M, Wacker HH, Burkhardt B, Harder L, et al. Diffuse large B-cell lymphoma in pediatric patients belongs predominantly to the germinal-center type B-cell lymphomas: a clinicopathologic analysis of cases included in the German BFM (Berlin-Frankfurt-Munster) Multicenter Trial. Blood. 2006;107(10):4047-4052.
doi pubmed - Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, Hutchinson A, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44(6):651-658.
doi pubmed - Dowiak AV, Tirado CA. Cytogenetic Characterization of Myeloid Neoplasms with t(2;3)(p13-25;q25-29): An Analysis of 60 Cases. J Assoc Genet Technol. 2017;43(2):64-69.
pubmed - Xing X, Lin D, Ran W, Liu H. ALK-positive diffuse large B-cell lymphoma of the duodenum: A case report and review of the literature. Exp Ther Med. 2014;8(2):409-412.
doi pubmed - Sibon D, Fournier M, Briere J, Lamant L, Haioun C, Coiffier B, Bologna S, et al. Long-term outcome of adults with systemic anaplastic large-cell lymphoma treated within the Groupe d'Etude des Lymphomes de l'Adulte trials. J Clin Oncol. 2012;30(32):3939-3946.
doi pubmed - Johnson NA, Savage KJ, Ludkovski O, Ben-Neriah S, Woods R, Steidl C, Dyer MJ, et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood. 2009;114(11):2273-2279.
doi pubmed - Stevens-Kroef M, Poppe B, van Zelderen-Bhola S, van den Berg E, van der Blij-Philipsen M, Geurts van Kessel A, Slater R, et al. Translocation t(2;3)(p15-23;q26-27) in myeloid malignancies: report of 21 new cases, clinical, cytogenetic and molecular genetic features. Leukemia. 2004;18(6):1108-1114.
doi pubmed - Shustik J, Han G, Farinha P, Johnson NA, Ben Neriah S, Connors JM, Sehn LH, et al. Correlations between BCL6 rearrangement and outcome in patients with diffuse large B-cell lymphoma treated with CHOP or R-CHOP. Haematologica. 2010;95(1):96-101.
doi pubmed - Zhao X, Fan R, Lin G, Wang X. Chromosome abnormalities in diffuse large B-cell lymphomas: analysis of 231 Chinese patients. Hematol Oncol. 2013;31(3):127-135.
doi pubmed - Antoniou AC, Wang X, Fredericksen ZS, McGuffog L, Tarrell R, Sinilnikova OM, Healey S, et al. A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor-negative breast cancer in the general population. Nat Genet. 2010;42(10):885-892.
doi pubmed - Paulsson K, Jonson T, Ora I, Olofsson T, Panagopoulos I, Johansson B. Characterisation of genomic translocation breakpoints and identification of an alternative TCF3/PBX1 fusion transcript in t(1;19)(q23;p13)-positive acute lymphoblastic leukaemias. Br J Haematol. 2007;138(2):196-201.
doi pubmed - Scholtysik R, Nagel I, Kreuz M, Vater I, Giefing M, Schwaenen C, Wessendorf S, et al. Recurrent deletions of the TNFSF7 and TNFSF9 genes in 19p13.3 in diffuse large B-cell and Burkitt lymphomas. Int J Cancer. 2012;131(5):E830-835.
doi pubmed - Calin GA, Croce CM. MicroRNAs and chromosomal abnormalities in cancer cells. Oncogene. 2006;25(46):6202-6210.
doi pubmed - Kim S, Kim H, Kang H, Kim J, Eom H, Kim T, Yoon SS, et al. Clinical significance of cytogenetic aberrations in bone marrow of patients with diffuse large B-cell lymphoma: prognostic significance and relevance to histologic involvement. J Hematol Oncol. 2013;6:76.
doi pubmed - Afonina IS, Elton L, Carpentier I, Beyaert R. MALT1—a universal soldier: multiple strategies to ensure NF-kappaB activation and target gene expression. FEBS J. 2015;282(17):3286-3297.
doi pubmed - Dierlamm J, Murga Penas EM, Bentink S, Wessendorf S, Berger H, Hummel M, Klapper W, et al. Gain of chromosome region 18q21 including the MALT1 gene is associated with the activated B-cell-like gene expression subtype and increased BCL2 gene dosage and protein expression in diffuse large B-cell lymphoma. Haematologica. 2008;93(5):688-696.
doi pubmed - Watanabe R, Tomita N, Matsumoto C, Hattori Y, Matsuura S, Takasaki H, Hashimoto C, et al. The 3q27 and 18q21 translocations for follicular lymphoma and diffuse large B-cell lymphoma in the rituximab era. J Clin Exp Hematop. 2013;53(2):107-114.
doi pubmed - Berglund M, Enblad G, Flordal E, Lui WO, Backlin C, Thunberg U, Sundstrom C, et al. Chromosomal imbalances in diffuse large B-cell lymphoma detected by comparative genomic hybridization. Mod Pathol. 2002;15(8):807-816.
doi pubmed - Yang Y, Kelly P, Shaffer AL, 3rd, Schmitz R, Yoo HM, Liu X, Huang DW, et al. Targeting non-proteolytic protein ubiquitination for the treatment of diffuse large B cell lymphoma. Cancer Cell. 2016;29(4):494-507.
doi pubmed - Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105(36):13520-13525.
doi pubmed - Toze CL, Barnett MJ, Naiman SC, Horsman DE. Trisomy 14 is a non-random karyotypic abnormality associated with myeloid malignancies. Br J Haematol. 1997;98(1):177-185.
doi pubmed - Guillaume B, Ameye G, Dierlamm J, Verhoef G, Duhem C, Ferrant A, Hagemeijer A, et al. Trisomy 16 as the sole anomaly in hematological malignancies. Three new cases and a short review. Cancer Genet Cytogenet. 2001;128(2):168-171.
doi - Liu W, Lyu R, Huang WY, Li CW, Liu H, Li J, Zou DH, et al. [Characteristics and prognostic significance of cytogenetic abnormalities in diffuse large b-cell lymphoma patients with bone marrow involvement]. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2017;25(3):761-765.
pubmed - Liberzon E, Avigad S, Yaniv I, Stark B, Avrahami G, Goshen Y, Zaizov R. Molecular variants of the ATM gene in Hodgkin's disease in children. Br J Cancer. 2004;90(2):522-525.
doi pubmed - Vick E, Patel K, Richardson N, et al. Age-Related Chromosomal Aberrations in Patients with Diffuse Large B-Cell Lymphoma. Blood. 2017;12(Suppl 1):1571–1571.
- Staton AD, Cohen JB. A clinician's approach to double-hit lymphoma: identification, evaluation, and management. J Oncol Pract. 2016;12(3):232-238.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.