

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 15, Number 3, June 2024, pages 472-481

Mitochondria of T Lymphocytes Promote Anti-Pulmonary Tumor Immune Response
Minsuk Kim
Department of Pharmacology, College of Medicine, Ewha Womans University, Seoul 07804, Korea
Manuscript submitted February 27, 2024, accepted March 30, 2024, published online April 15, 2024
Short title: Tumoricidal Mitochondria From Non-Activated T Cells
doi: https://doi.org/10.14740/wjon1841
| Abstract | ▴Top |
Background: B-cell lymphoma 2 (Bcl-2), a protein involved in apoptosis, has been proven to have carcinogenic potential and is well documented. With the recent advancement in optical technology, it has become possible to observe subcellular organelles such as mitochondria in real-time without the need for staining. Consequently, we have examined the movement of mitochondria in cancer cells, correlating it with the regulation of Bcl-2.
Methods: Using a tomographic microscope, which can detect the internal structure of cells, we observed lung tumor cells. Cells were exposed to a laser beam (λ = 520 nm) inclined at 45°, and holographic images were recorded up to a depth of 30 µm of reconstruction.
Results: Intriguingly, lung tumor cells rapidly expelled mitochondria upon the attachment of Bcl-2 or B-cell lymphoma extra-large (Bcl-xL) inhibitors. On the other hand, we observed that tumor cells hijack mitochondria from T cells. The hijacked mitochondria were not immediately linked to tumor cell death, but they played a role in assisting granzyme B-induced tumor cell death. Due to lower levels of Bcl-2 and Bcl-xL on the mitochondria of T cells compared to lung tumor cells, immune cells depleted of Bcl-2 and Bcl-xL were co-cultured with the tumor cells.
Conclusions: As a result, a more effective tumor cell death induced by granzyme B was observed. Additionally, further enhanced anticancer immune response was observed in vivo. Together, we show that modified mitochondria of T cells can provide potential novel strategies towards tumor cell death.
Keywords: Mitochondria; Tunneling nanotube; Pulmonary fibroblast tumor; Tomographic microscope; Refractive index
| Introduction | ▴Top |
There are many lung cancer treatments in development with various mechanisms [1-5]. Methotrexate suppresses cell division by inhibiting production of tetrahydrofolate, a necessary base for nucleic acid synthesis; 5-fluorouracil suppresses proliferation by DNA base substitution; bleomycin damages DNA and RNA in the G2 phase of the cell; topotecan inhibits topoisomerase, which is involved in DNA replication [6-8]. Following drug development, lung cancers have also been categorized into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) [9]. NSCLC represents 80% to 85% of all lung cancers [9]. NSCLC is characterized by mutations in epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), c-ros oncogene 1 (ROS1), and B-Raf proto-oncogene (BRAF) [10, 11]. Alectinib and ceritinib, which are ALK tyrosine kinase inhibitors, have currently been shown to be effective against NSCLC [12]. Eventually, the role of predictive biomarkers has been extensively studied in cancer patients [13-16]. As a result of their development, pembrolizumab, Opdivo, or nivolumab demonstrate remarkable efficacy. They prevent the binding of programmed cell death 1 (PD-1), thereby enhancing the immune response of T cells when interacting with lung tumor cells, leading to the destruction of tumor cells [17]. A systematic review and meta-analysis in patients with solid tumors demonstrated that immunotherapy, either alone or in combination, reduces the risk of death or progression [13]. However, even with targeted immunotherapies, complete eradication of lung tumor is still not attained.
B-cell lymphoma 2 (Bcl-2) is a protein originally discovered in B-cell lymphoma, and it can promote the proliferation of cancer cells through inhibiting cell death and enhancing cell survival [18, 19]. As a member of the Bcl-2 family of proteins, B-cell lymphoma extra-large (Bcl-xL) plays also a similar role to Bcl-2, exerting anti-apoptotic effects and promoting cell survival [20]. Bcl-2 and Bcl-xL have been known to be present in relatively high levels not only in lymphomas but also in lung cancer cells [2, 21]. Bcl-2 and Bcl-xL are located in the mitochondria, and they block the release of pro-apoptotic signals from mitochondria, thereby preventing cell death [22]. Therefore, Bcl-2 and Bcl-xL inhibitors have emerged as potential drug targets for cancer treatment. These proteins commonly possess a Bcl-2 homology 3 (BH3) domain, which is involved in the regulation of cell death [23]. Consequently, many BH3-mimetics are in development as anticancer drugs [24]. Venetoclax (ABT-199), navitoclax (ABT-263) or obatoclax are BH3-mimetics that specifically inhibit Bcl-2 [25, 26]. Therefore, mitochondrial Bcl-2 and Bcl-xL can provide important clues in our understanding of tumor cell fate. In order to gain a more detailed understanding of mitochondrial dynamics related to apoptosis in lung tumor, optical tomography was employed.
In optical tomography, images are acquired by capturing waves that pass through cells from different angles. Subsequently, digital reconstruction is performed to visualize the internal structure of cells in a cross-sectional manner [27-29]. A tomographic microscope can generate reconstructed cell images based on varying refractive index, allowing the detection of transparent structures such as mitochondria without the need for staining.
In this study, we utilized tomographic microscopy to analyze pulmonary tumor cells and their reaction to anti-tumor agents. Intriguingly, our findings revealed a transfer of mitochondria from immune cells to tumor cells, leading to apoptosis induced by granzyme B. This process resulted in a higher rate of cell death compared to the use of BH3 mimetics. Consequently, we hypothesized that intercepted mitochondria have the potential to enhance tumor apoptosis. To further explore this hypothesis, we delved into the mechanisms by which immune cell mitochondria can initiate apoptosis of tumor using HCC1195, HCC1438, or Lewis lung carcinoma (NSCLC models).
| Materials and Methods | ▴Top |
In vivo animal study
All mice (C57BL/6) were purchased from Charles River. Animal experiments were approved by the Ewha Womans University Animal Care Committee (Institutional Review Board (IRB) number: ESM14-0260). Forty male mice (about 20 g; 8 - 10 weeks) were subcutaneously injected with Lewis lung cancer cells (1 × 106 cells). Each animal was intraperitoneally injected every alternate day (eight cycles) with anti-immunoglobulin (Ig)G2a, anti-PD-1 (10 mg/kg), T cells (1 × 106 cells), or Bcl-2 knocked out (KO) T cells (1 × 106 cells). Blood was collected from the mouse, and CD69-positive cells were isolated using the CD69 kit II (130-092-355, Miltenyi Biotec, Germany). The activated T cells were then quantitatively measured using the CD69 enzyme-linked immunoassay (ELISA) kit (EH96RB, ThermoFisher, USA).
Cell culture
Human lung cancer lines, HCC1195 (ATCC; RRID: CVCL_5127) or HCC1438 (ATCC; RRID: CVCL_L088) were maintained in Roswell Park Memorial Institute Medium 1640 (RPMI 1640) (11875093, Thermofisher, USA) with 10% bovine serum (16000044, Gibco, USA) at 37 °C under an atmosphere of 95% O2 and 5% CO2. The cells were treated with ABT199 (3 nM, 6960, Tocris, UK), A115463 (19 nM, S7800, Selleck Chemicals, USA), or ABT737 (15 µM, 6835, Tocris, UK) for 1 h. Apoptosis was induced following combinations of recombinant perforin (100 pg/µL, ENZ-PRT313-0010, Enzo Life Science, USA) and granzyme B (200 pg/µL, 10345-H08H, Sinobiological, China). CaCl2 (1.25 mM) was added for perforin activity. Human CD3+ T cells (PB03020C, StemExpress, USA) were obtained through the protocol number 401-01 and the IRB tracking number 20152869. T cells were cultured in AIM V medium (12055091, Thermo Fisher Scientific, USA), supplemented with 10% fetal calf serum (A3520502, Thermo Fisher Scientific, USA). Human pulmonary fibroblast (C-12360, PromoCell, Germany) were cultured in fibroblast growth medium 2 (C-23020, PromoCell, Germany).
Isolation of mitochondrial fraction
Cells were collected using homogenization buffer (70 mM sucrose, 220 mM mannitol, and 20 mM HEPES) containing a protease inhibitor (89874, Thermofisher, USA) and centrifuged at 2,300 g for 5 min. The pellet was suspended in homogenization buffer on ice for 5 min. Cell rupture was achieved with 10 strokes using a 27-gauge needle. The homogenate was then centrifuged at 400 g for 5 min. Following a subsequent centrifugation at 5,800 g for 5 min, mitochondrial fractions were collected and suspended in homogenization buffer. The quantity of isolated mitochondria was determined by measuring protein concentration using the Bio-Rad protein assay kit (Bio-Rad, Richmond, USA).
The de novo synthesis of ABT199-boron dipyrromethene (BODIPY)
A procedure for synthesis of ABT199-BODIPY is summarized as follows [30]. Sulfonamide analog was synthesized via nucleophilic aromatic substitution of commercially available 4-fluoro-3-nitrobenzenesulfonamide with tertbutyl 4-(aminomethyl) piperidine-1-carboxylate. Removal of the tert-butoxycarbonyl protecting group with HCl in dioxane followed by alkylation with 1-N-(2-bromoethyl) carbamate afforded the sulfonamide analog. The tert-butoxycarbonyl protecting group was removed using trifluoroacetic acid and Et3SiH (as a scavenger) followed by reaction with the N-hydroxysuccinimidyl ester of BODIPY FL to afford ABT-199-BODIPY. The purity was 95% based on liquid chromatography-mass spectrometry (LC-MS) analysis and ultraviolet-visible (UV-Vis) detection.
Optical tomography
A green laser (λ = 520 nm, exposure 0.2 mw/mm2) was split into cells at three-dimensional (3D) explorer (Nanolive, Switzerland) [27]. Cells were exposed to a laser beam inclined at 45°. Holographic images were captured with the reference beam. 3D cell images were recorded up to a depth of 30 µm of reconstruction. Mitochondria were stained using Mitotracker (M7514, Thermofisher, USA) at 490 nm.
Western blot
Briefly, cells (0.4 × 106) were homogenized in ice-cold lysis buffer. After centrifugation at 5,000 g for 30 min, the protein content of the supernatant was quantified using a Bradford protein assay. Samples were diluted, boiled with sample loading dye, and 100 µg were used in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (4561033EDU, Bio-Rad, USA). After blotting, membranes were blocked in 5% skim milk (70,166, Sigma-Aldrich) in tris-buffered saline containing 0.1% Tween-20 (P1379, Sigma-Aldrich). Membranes were incubated with Bcl-2 (1:1,000; ab59348, RRID: AB_289591, Abcam, UK), Bcl-xL (1:1,000; ab32370, RRID: AB_2847960, Abcam, UK), cytochrome c oxidase subunit IV (COX IV) (1:1,000; ab202554, RRID: AB_2847960, Abcam, UK), Bid (1:1,000; ab32060, RRID: AB_365478, Abcam, UK), cytochrome C (1:1,000; #11,940, RRID: AB_2637071, Cell signaling technology, USA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1,000; ab8245, RRID: AB_1267174, Abcam, UK) or beta-tubulin (1:1,000; ab179513, RRID: AB_1566837, Abcam, UK), and subsequently with secondary goat anti-rabbit horseradish peroxidase-conjugated antibody. Reaction products were visualized using an enhanced chemiluminescence detection kit (32106, Thermo Scientific, USA) and quantified by densitometry.
Detection of apoptosis or caspase-3 activity
Apoptosis was determined using ELISA kit (APT225, Sigma, USA). Briefly, cells were fixed with 2,2’-azino-bis-3-ethylbenzothiazoline-6-sulfonic acid (ABTS) for 30 min at 37 °C. To denature DNA, cells were incubated for 20 min at 75 °C. After cooling at 4 °C for 5 min, plates were blocked in 5% skim milk (70,166, Sigma, USA) in phosphate-buffered saline at 37 °C for 1 h. Cells were incubated with antisera mixture and washed with phosphate-buffered saline at 37 °C for 1 h. After treatment with stop solution, absorbance was measured at 405 nm. To determine caspase-3 activity, cells were fixed and incubated with antibody cocktail or tetramethylbenzidine (TMB) at 37 °C for 1 h. After treatment with stop solution, the degree of activation of relative was measured at 450 nm.
Wild type transfection or gene deletion using clustered regularly interspaced short palindromic repeats (CRISPR)
CRISPR or wild type transfection of Bcl-2 and Bcl-xL in human lung cancer cells was performed using a kit from Santa Cruz (sc-395739, Santacruz, USA). Briefly, in six-well culture plates, 106 cells were plated and exposed to the Bcl-2 wild type plasmid (8768, RRID: Addgene_8768, Addgene, USA), Bcl-xL wild type plasmid (113458, RRID: Addgene_113458, Addgene, USA), Bcl-2 CRISPR plasmid (sc-400025-KO-2, Santacruz, USA), Bcl-xL CRISPR plasmid (sc-400170, Santacruz, USA) or control plasmid (sc-418922, Santacruz, USA) solution for 8 h at 37 °C in a CO2 incubator. Then, media was changed to Dulbecco modified eagle medium (DMEM) with 10% bovine serum and incubated for another 12 h.
Statistical analysis
Values are means ± standard error (SE). Wherever appropriate, one-way analysis of variance (ANOVA) followed by the Bonferroni test was used to determine differences between group mean values. The level of statistical significance was set at P < 0.05.
All human/animal studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
| Results | ▴Top |
Poor tumoricidal activity of BH3-mimetics in pulmonary tumor cells
Bcl-2 and Bcl-xL have been known to be highly expressed in pulmonary tumor cells [31]. They are more predominantly concentrated in mitochondria compared to the cytosol in HCC1195 or HCC1438 lung tumor cell lines (Fig. 1a-c). To block Bcl-2 and Bcl-xL, BH3-mimetics such as ABT199, A115463, and ABT737 were used (Fig. 1d-f). ABT199 is a selective Bcl-2 inhibitor, while A115463 is designed to inhibit Bcl-xL, and ABT737 inhibits both Bcl-2 and Bcl-xL [32, 33]. HCC1195 or HCC1438 cell lines were treated with BH3-mimetics, but no significant tumor cell death was observed. To induced additional cell death, they were co-treated with granzyme B and perforin. Treatment with granzyme B and perforin resulted in 16% induction of cell death, but BH3-mimetics did not show any additional tumor cell death. Treatment with BH3-mimetics at a three-fold higher concentration induced approximately 20% of the tumor cell death (Supplementary Material 1, www.wjon.org). However, it also resulted in 30% cell death in pulmonary fibroblasts. This was considered to be due to the toxicity of the chemical substance itself rather than the tumoricidal effects of BH3-mimetics. To track BH3-mimetics in tumor cells, BODIPY was attached to ABT199 (Fig. 1g) [30]. After treatment of ABT199-BODIPY in HCC1195 cells, cells were monitored using a tomographic microscopy (Fig. 1h, i). This process was scanned with a λ = 520 nm, and the holographic images were subsequently recorded. Intriguingly, ABT199 remained intracellularly for 1 h in pulmonary fibroblasts, whereas about half of them disappeared within 15 min, and a negligible concentration was present intracellularly within 1 h in HCC1195 or HCC1438 cells. In summary, we found that the poor antitumor activity of BH3-mimetics was likely due to their quick elimination from HCC1195 or HCC1438 cells.
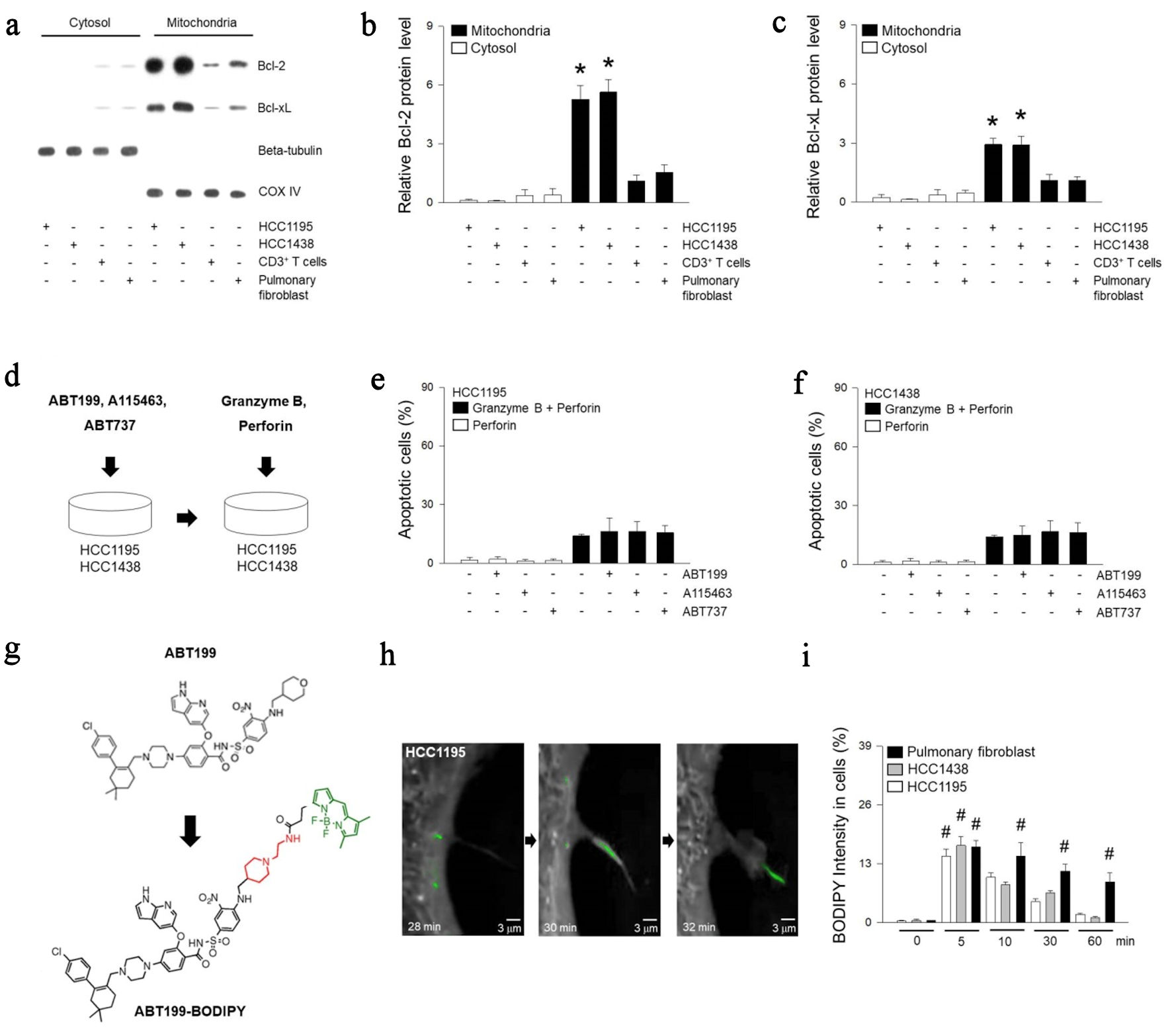 Click for large image | Figure 1. Treatment of BH3-mimetic drugs in HCC1195 and HCC1438 cells. (a-c) Cytosol and mitochondrial proteins were extracted from HCC1195, HCC1438, CD3+ T, pulmonary fibroblast cells, and expression of Bcl-2 or Bcl-xL was determined using western blotting. Beta-tubulin was used as a positive marker for the cytosol, while COX IV was used as a positive marker for the mitochondrial fraction. (d) Cell death was measured following treatment with 3 nM ABT199, 19 nM A115463, or 15 µM ABT737 for 1 h, or incubation with 200 pg/µL granzyme B and 100 pg/µL perforin for 2 h. (e, f) The cells were fixed up to 5 h, and apoptosis were determined using ELISA kit. (g) Structures of ABT199 and ABT199-BODIPY. (h) Following treatment of synthetic ABT199-BODIPY, refractive index images of HCC1195 were enlarged at 28, 30, or 32 min, and ABT199-BODIPY were colored in green. (i) Quantified mean fluorescent intensity of the BODIPY signal in cells was measured at 512 nm. Results are the means ± standard error (SE) of six experiments in each group. *Significantly different from cytosol fraction, P < 0.05. #Significantly different from treatment of ABT199-BODIPY at 0 min, P < 0.05. COX IV: cytochrome c oxidase subunit IV; BH3: Bcl-2 homology 3; Bcl-2: B-cell lymphoma 2; Bcl-xL: B-cell lymphoma extra-large; ELISA: enzyme-linked immunoassay; BODIPY: boron dipyrromethene. |
Mitochondrial migration of T cells promotes tumor cell death by granzyme B
It has been reported that various tumor cells hijack mitochondria from non-activated T cells [34]. Non-activated T cells were stained with mitotracker and cultured together with HCC1195, HCC1438 or pulmonary fibroblasts (Fig. 2a, b). Using tomographic microscopy, we observed that HCC1195 hijacked mitochondria of T cells. Using fluorescence microscopy, we confirmed that the stained mitochondria of the immune cells migrated to HCC1195 or HCC1438 (Fig. 2c, d). This appeared to be specific to tumor cells, as pulmonary fibroblasts did not show any significant mitochondrial migration. The mitochondrial movement itself did not affect apoptosis. However, the mitochondrial transfer promoted granzyme B-mediated cell death of the pulmonary tumor cells (Fig. 2e-g).
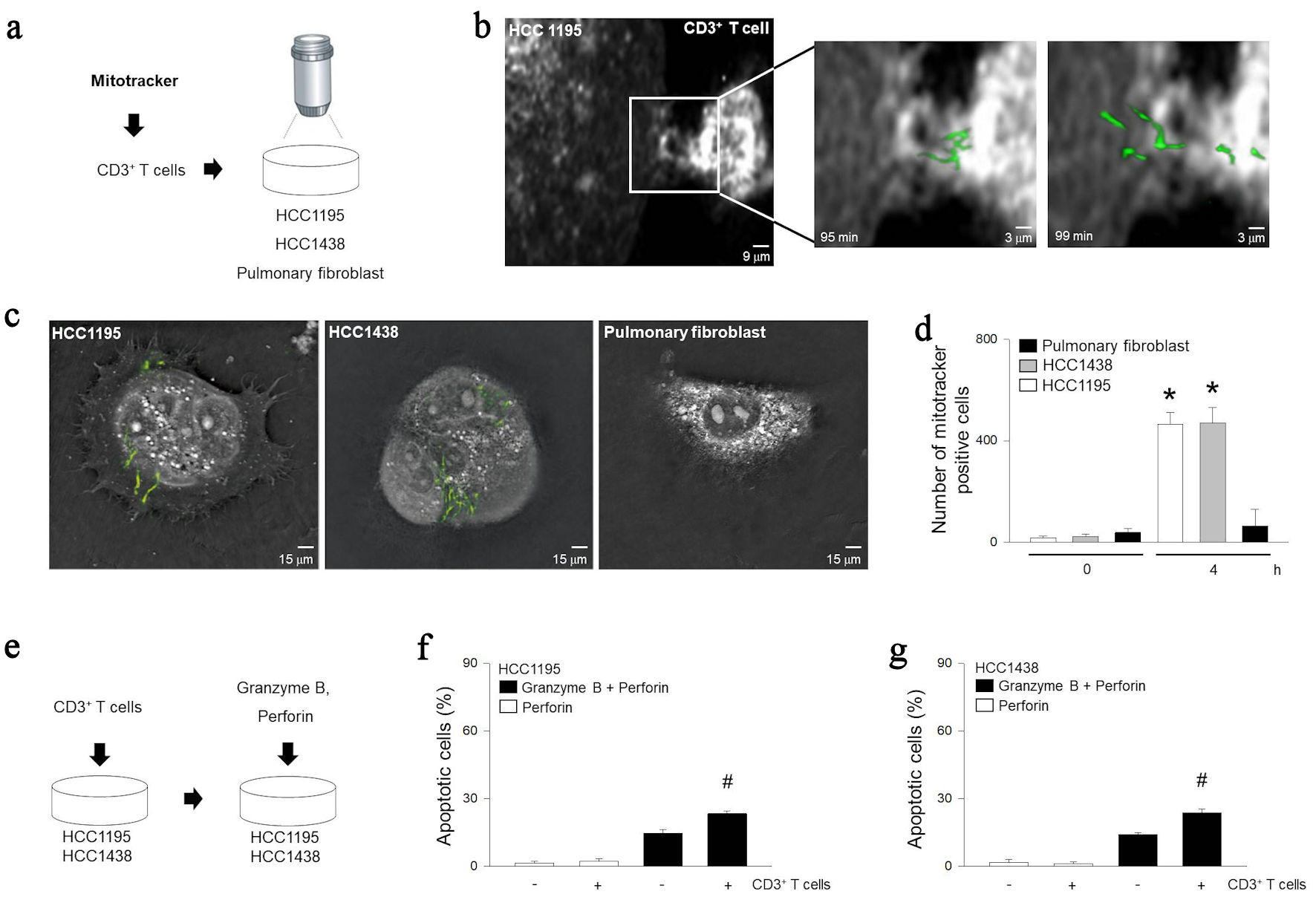 Click for large image | Figure 2. Relationship between granzyme B-induced apoptosis and mitochondrial trafficking from immune cells. (a) Diagram for observing mitochondrial internalization. After treating inactivated T cells with mitotracker and incubating with HCC1195, HCC1438, or pulmonary fibroblast for 5 h. (b) Cell images were rendered with tomographic microscopy. (c) Cellular internalization of mitochondria was visualized with mitotracker at 490 nm. (d) Quantization of internalization of mitochondria was determined by tomographic phase microscopy. (e) Diagram for model of apoptosis with mitochondrial trafficking from immune cells. After incubating inactivated T cells with HCC1195 or HCC1438 cells for 5 h, apoptosis was induced following combinations of recombinant 100 pg/µL of perforin and 200 pg/µL of granzyme B. (f, g) Apoptosis was determined using ELISA kit. Results are the means ± standard error (SE) of six experiments in each group. *Significantly different from incubation of CD3+ T cells at 0 h, P < 0.05. #Significantly different from treatment of granzyme B and perforin, P < 0.05. ELISA: enzyme-linked immunoassay. |
Role of transmigrated mitochondrial Bcl-2 and Bcl-xL in pulmonary tumor cells
To understand the role of mitochondrial proteins in T cells, Bcl-2 and Bcl-xL were KO or overexpressed (OE) in T cells and designated as KO and OE respectively (Fig. 3a, b). KO or OE of Bcl-2 or Bcl-xL did not induce T cell death (Fig. 3c). However, tumor cells showed blebbing and microtubule spikes, demonstrating increased apoptosis in KO T cells in response to granzyme B (Fig. 3d, e). Mitochondria of KO or OE T cells migrated to tumor cells in a similar manner (Fig. 3f). However, OE T cells did not induce additional tumoricidal effects in response to granzyme B (Fig. 3g, h).
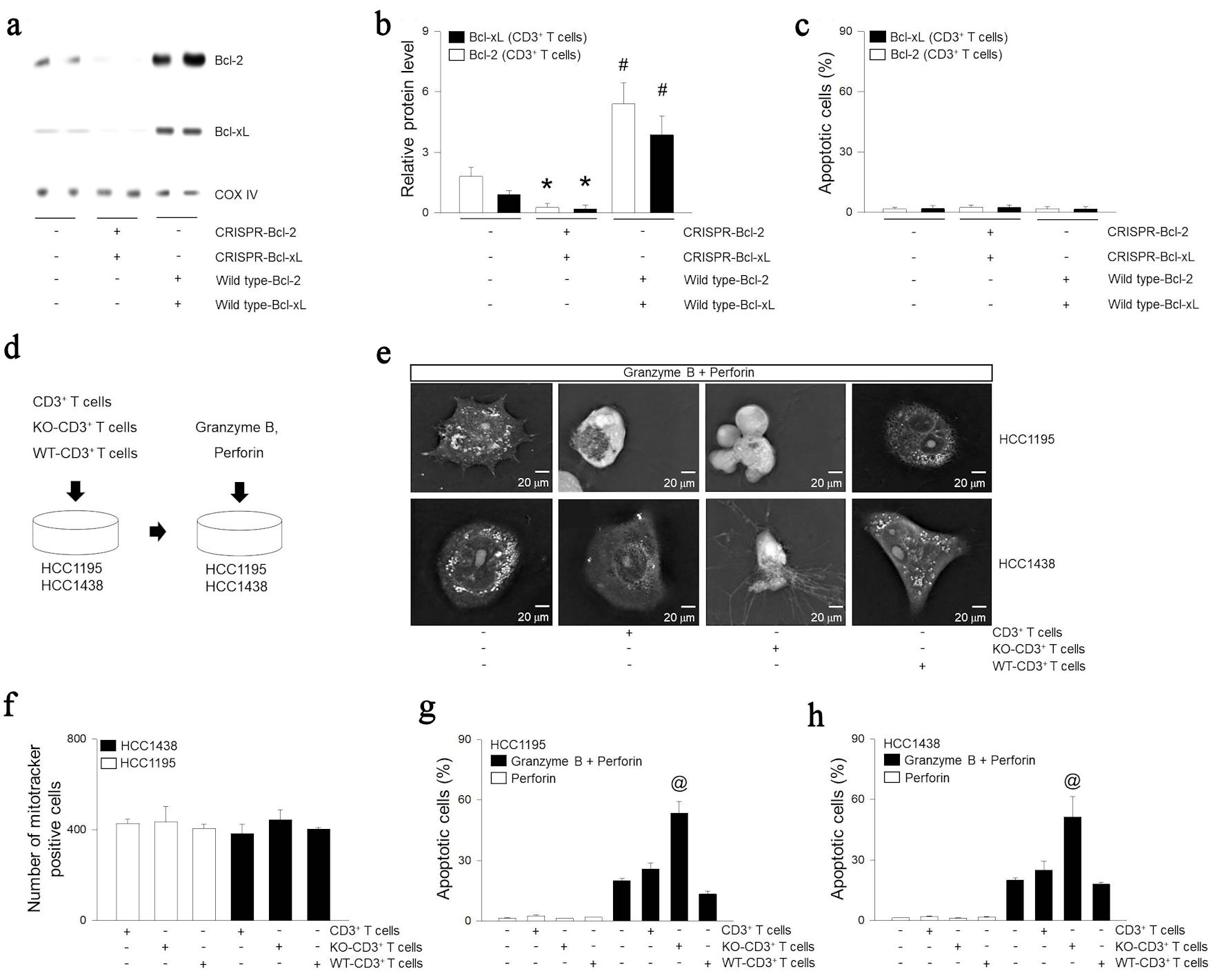 Click for large image | Figure 3. Role of expression of Bcl-2 or Bcl-xL in extracellular mitochondria on lung cancer cells. (a, b) CD3+ T cells treated with Bcl-2 CRISPR-plasmid, Bcl-2 wild type (WT)-plasmid, Bcl-xL CRISPR-plasmid, or Bcl-xL wild type-plasmid. The overexpressed (OE) and knocked out (KO) indicate overexpress and knock out. Mitochondrial proteins were extracted from CD3+ T cells and expressions of Bcl-2, Bcl-xL, or COX IV were determined using western blotting. (c) Apoptosis was determined using ELISA kit. (d) CD3+ T cells, KO-CD3+ T cells, or OE-CD3+ T cells was incubated with HCC1195 or HCC1438 cells for 5 h. Apoptosis was induced following combinations of recombinant 100 pg/µL of perforin and 200 pg/µL of granzyme B. Cell images were rendered (e) or quantization of internalization of mitochondria (f) was determined by tomographic phase microscopy. (g, h) Apoptosis was determined using ELISA kit. Results are the means ± standard error (SE) of six experiments in each group. *Significantly different from treatment of negative control plasmid, P < 0.05. #Significantly different from treatment of CRISPR plasmid, P < 0.05. @Significantly different from all other groups, P < 0.05. Bcl-2: B-cell lymphoma 2; Bcl-xL: B-cell lymphoma extra-large; COX IV: cytochrome c oxidase subunit IV; CRISPR: clustered regularly interspaced short palindromic repeats; ELISA: enzyme-linked immunoassay. |
Bcl-2 and Bcl-xL KO mitochondria enhance cytochrome C-induced apoptosis
We observed BH3 interacting domain death agonist (Bid) was truncated (tBid) in both HCC1195 and HCC1438 following granzyme B treatment (Fig. 4a-c). The entry of Bcl-2 and Bcl-xL KO mitochondria did not alter the total amount of Bcl-2 and Bcl-xL proteins in HCC1195 and HCC1438 cells (Fig. 4d, e). However, infiltration of Bcl-2 and Bcl-xL KO mitochondria with granzyme B led to increase cytochrome C in cytosol and caspase-3 activity in HCC1195 and HCC1438 cells (Fig. 4f, g).
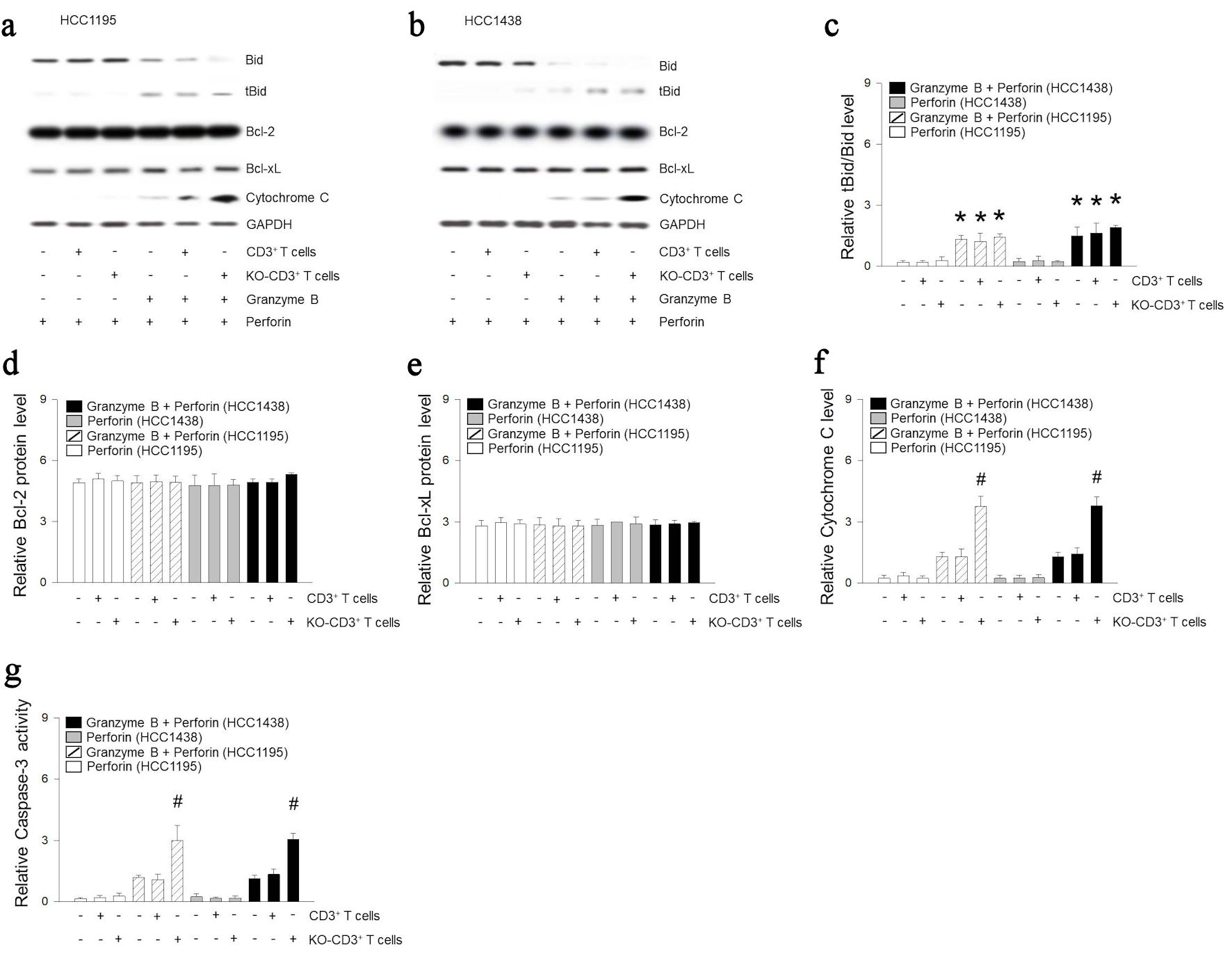 Click for large image | Figure 4. Mitochondria of immune cells deficient in Bcl-2 and Bcl-xL are related to cytochrome C-related apoptotic signals. (a-f) CD3+ T cells was incubated with HCC1195 or HCC1438 cells for 5 h. Apoptosis was induced following combinations of perforin and granzyme B. Proteins were extracted from HCC1195 or HCC1438 cells, and expression of Bid, Bcl-2, Bcl-xL, cytochrome C or GAPDH were determined using western blotting. (g) Caspase-3 activity was determined using ELISA kit. Results are the means ± standard error (SE) of six experiments in each group. *Significantly different from treatment of perforin, P < 0.05. #Significantly different from incubation of CD3+ T cells, P < 0.05. Bcl-2: B-cell lymphoma 2; Bcl-xL: B-cell lymphoma extra-large; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; ELISA: enzyme-linked immunoassay; KO: knocked out. |
Mitochondrial hijacking enhances the anti-tumor immune response in vivo
To test for mitochondrial hijack by cancer cells in vivo, we injected Lewis lung carcinoma cells into C57BL/6 mice (Fig. 5a). After 8 days, we injected T cells or anti-PD-1 for 12 days. Anti-PD-1 injection did not alter the proportion of activated T cells (Fig. 5b). T cells or KO T cells treatment did not show change in tumor volume (Fig. 5c, d). In contrast, anti-PD-1 reduced tumor volume by 13%. Moreover, KO T cell treatment with anti-PD-1 induced an additional 28% reduction in tumor volume. In summary, migration of Bcl-2 and Bcl-xL KO mitochondria resulted in potentiation of tumoricidal effects of activated T cells (Fig. 5e).
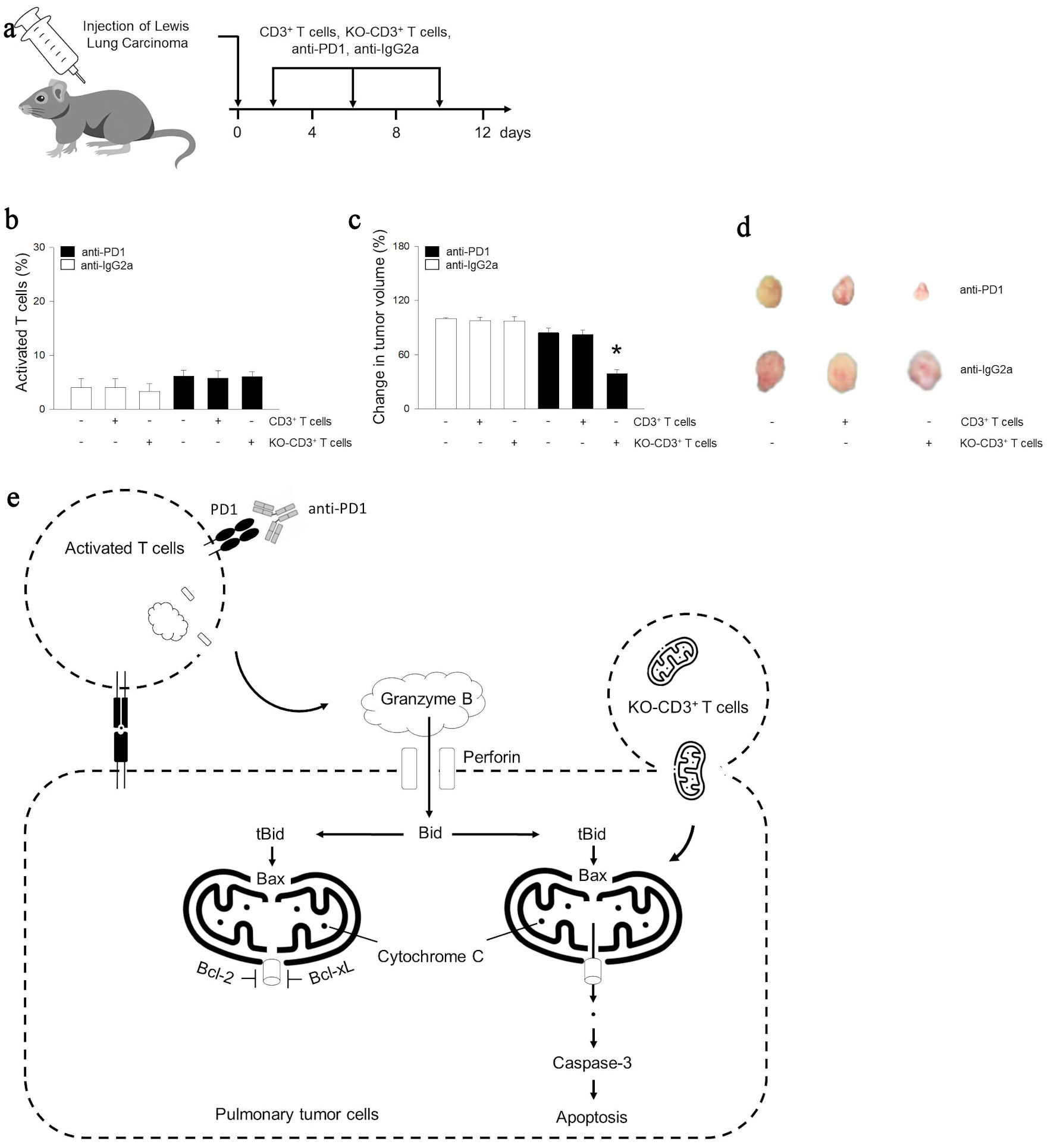 Click for large image | Figure 5. Mitochondria of immune cells prevents tumor growth following activated immunity in vivo. (a) Beginning at 5 weeks of age, male mice were given intraperitoneally with anti-IgG2a, anti-PD-1 (10 mg/kg), CD3+ T cells (1 × 106 cells), or KO-CD3+ T cells (1 × 106 cells) for 12 days. The arrow indicates the injection days. (b) Graph showing the effect of treatments on the population of activated CD69+ T cells using CD69 ELISA kit. (c, d) Bar graph or images of tumor volume change for mice. (e) Cellular internalization of mitochondria from immune cells promoted antitumor immune response through cytochrome C related apoptosis. Results are the means ± standard error (SE) of six experiments in each group. *Significantly different from all other groups, P < 0.05. ELISA: enzyme-linked immunoassay; KO: knocked out; Bcl-2: B-cell lymphoma 2; Bcl-xL: B-cell lymphoma extra-large; ELISA: enzyme-linked immunoassay; Ig: immunoglobulin; PD-1: programmed cell death 1. |
| Discussion | ▴Top |
Granzyme B is a caspase-like serine protease that is released by cytotoxic T cells to kill tumor cells [35]. It induces truncated Bid to secrete cytochrome C from mitochondria [36]. Cytochrome C in turn activates caspase-3 to initiate the apoptotic pathway [37]. However, Bcl-2 and Bcl-xL block the release of cytochrome C from the mitochondria, thereby preventing apoptosis [38]. Therefore, Bcl-2 and Bcl-xL regulate cell death by localizing in the mitochondrial membrane [22]. We observed that Bcl-2 and Bcl-xL were predominant in the mitochondria rather than in the cytoplasm in HCC1195 and HCC1438 cells compared to pulmonary fibroblasts. To investigate the role of Bcl-2 and Bcl-xL in apoptosis of pulmonary tumor cells, BH3-mimetic ABT199, A115463, and ABT737 were tested. These had insignificant tumoricidal effects. Use at a concentration three times the therapeutic dose led to tumor cell death, but this led to further apoptosis of pulmonary fibroblasts, suggesting loss of tumoricidal effects.
Recently, it has been reported that tumor cells hijack the mitochondria of non-activated T cells [34]. Mitochondrial migration was also observed in HCC1195 and HCC1438 cells using tomographic microscopy. However, the mitochondrial migration itself did not induce the tumor cell death. To recapitulate a scenario in which activated T cells attack tumor cells, HCC1195 and HCC1438 were treated with granzyme B and perforin. An increase in tBid was observed, but cytochrome C was not released from mitochondria following granzyme B. However, hijacked mitochondria of immune cells led to release of cytochrome C resulting in apoptosis in response to granzyme B. We postulated this is likely due to the relatively low abundance of Bcl-2 and Bcl-xL in the hijacked mitochondria. To test this hypothesis, Bcl-2 and Bcl-xL were knocked out in T cells and co-cultured with HCC1195 or HCC1438 cells. The entry of Bcl-2 and Bcl-xL KO mitochondria did not alter the total amount of Bcl-2 and Bcl-xL proteins in HCC1195 and HCC1438 cells. This suggests that the predominant proportion of Bcl-2 and Bcl-xL are from tumor cells. However, the entry of KO mitochondria significantly increased the cytochrome C release to the cytoplasm and enhanced caspase-3 activation resulting in blebbing and microtubule spikes in response to granzyme B in tumor cells, indicating tumor cell apoptosis. To evaluate the role of the process in vivo, mice with Lewis lung carcinoma were treated with either anti-PD-1 or KO T cells. The treatments did not alter the amount of activated T cells. The administration of anti-PD-1 alone resulted in a tumor volume reduction of approximately 13%. However, combined treatment of with KO T cells demonstrated an additional 28% reduction in the tumor volume.
Following the signaling of cell death, the expectation is often that tumor cells will undergo apoptosis. However, the experimental results are often below expectations. Through this study, it became evident that the presence of normal mitochondria capable of secreting cytochrome C is essential for cell death. The potential of this study lies in changing perceptions regarding mitochondria. In fact, conventional biology textbooks typically depict mitochondria as being present in small quantities within cells. However, it has been revealed that mitochondria, shaped like spaghetti, occupy every nook and cranny of the cell [39]. Given the abundance of mitochondria within cells, it is somewhat reasonable for tumor cells to expel mitochondria that are bound to chemicals such as Bcl-2. Conversely, the presence of mitochondria not encircled by Bcl-2 appears to actively support tumor cell death, as partially confirmed by this experiment. The prevailing biased perception of mitochondria likely delayed such discoveries, and it is imperative to focus on improving perceptions by reporting continued discoveries of mitochondrial activities, shapes, and mobility over the next 5 years.
Limitations of this research
In fact, a more suitable model for investigating Bcl-2 deficiency would involve delivering mitochondria that cannot bind to Bcl-2 into tumor cells. However, such mitochondria have not been studied or developed. Furthermore, immune cells without the assistance of anti-PD-1 may not simply abandon tumor cells but could potentially enhance the apoptotic potential of cancer cells by delivering normal mitochondria. Nonetheless, despite these considerations, the abundance of Bcl-2 expression within cancer cells may lead to the progressive encirclement of normal mitochondria by Bcl-2 over time, potentially weakening the apoptotic function of the cells. However, this could serve as an explanatory clue as to how cancer cells acquire resistance to immunotherapy. Many questions and hypotheses regarding cancer cell mitochondria may arise, but the lack of experimental tools and methods of mitochondria to address these questions remains a limitation of this study.
Conclusions
In summary, our study revealed that knockout of Bcl-2 and Bcl-xL in mitochondria enhances the apoptotic potential of pulmonary tumor cells. Our findings suggest that modified mitochondria from T lymphocytes can be utilized to promote apoptosis in pulmonary tumor cells following PD-1 blockade.
| Supplementary Material | ▴Top |
Suppl 1. (A-C) Cell death was measured following treatment with 3 nM ABT199, 19 nM A115463, or 15 µM ABT737 for 1 h, or incubation with 9 nM ABT199, 57 nM A115463, or 45 µM ABT737 for 1 h. The Cells were fixed up to 5 h, and apoptosis were determined using ELISA kit. Results are the means ± SE of six experiments in each group. *Significantly different from optimal concentration treatment of BH3-mimetic drugs, P < 0.05.
Acknowledgments
We thank Minjeong Kim (Ewha medical student) for helping to culture tumor cells.
Financial Disclosure
This work was supported by the National Research Foundation of Korea (NRF) grant (2020R1A5A2019210 and 2019R1C1C1003384) funded by the Korean government (MSIT), Ewha Womans University Research Grant of 2023.
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
All informed consent for all human cell research was obtained after IRB approval, and informed consent was obtained from the donors. All donors consented to the publication of the research purposes and outcomes.
Author Contributions
MK conceptualized and designed, performed experiments, analyzed data, prepared figures and wrote manuscript. The author read and approved the final manuscript.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Guo Q, Liu L, Chen Z, Fan Y, Zhou Y, Yuan Z, Zhang W. Current treatments for non-small cell lung cancer. Front Oncol. 2022;12:945102.
doi pubmed pmc - Alam M, Alam S, Shamsi A, Adnan M, Elasbali AM, Al-Soud WA, Alreshidi M, et al. Bax/Bcl-2 cascade is regulated by the EGFR pathway: therapeutic targeting of non-small cell lung cancer. Front Oncol. 2022;12:869672.
doi pubmed pmc - Ikezawa Y, Mizugaki H, Morita R, Tateishi K, Yokoo K, Sumi T, Kikuchi H, et al. Current status of first-line treatment with pembrolizumab for non-small-cell lung cancer with high PD-L1 expression. Cancer Sci. 2022;113(6):2109-2117.
doi pubmed pmc - Suryavanshi SV, Kulkarni YA. NF-kappabeta: a potential target in the management of vascular complications of diabetes. Front Pharmacol. 2017;8:798.
doi pubmed pmc - Rocco D, Malapelle U, Del Re M, Della Gravara L, Pepe F, Danesi R, Troncone G, et al. Pharmacodynamics of current and emerging PD-1 and PD-L1 inhibitors for the treatment of non-small cell lung cancer. Expert Opin Drug Metab Toxicol. 2020;16(2):87-96.
doi pubmed - Sekimura A, Iwai S, Funasaki A, Motono N, Usuda K, Uramoto H. Lung cancer combined with methotrexate-associated lymphoproliferative disorder: a case report. Int J Surg Case Rep. 2019;59:161-164.
doi pubmed pmc - Zhao L, Wang WJ, Zhang JN, Zhang XY. 5-Fluorouracil and interleukin-2 immunochemotherapy enhances immunogenicity of non-small cell lung cancer A549 cells through upregulation of NKG2D ligands. Asian Pac J Cancer Prev. 2014;15(9):4039-4044.
doi pubmed - Stathopoulos GP, Katis C, Tsavdaridis D, Dimitroulis J, Karaindros D, Stathopoulos J, Dimou E. Front-line paclitaxel and topotecan chemotherapy in advanced or metastatic non-small-cell lung cancer: a phase II trial. Cancer Chemother Pharmacol. 2006;58(4):555-560.
doi pubmed - Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893-1907.
doi - Du X, Shao Y, Qin HF, Tai YH, Gao HJ. ALK-rearrangement in non-small-cell lung cancer (NSCLC). Thorac Cancer. 2018;9(4):423-430.
doi pubmed pmc - Doebele RC, Camidge DR. Targeting ALK, ROS1, and BRAF kinases. J Thorac Oncol. 2012;7(16 Suppl 5):S375-376.
doi pubmed pmc - Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189-1197.
doi pubmed pmc - Mollica V, Rizzo A, Marchetti A, Tateo V, Tassinari E, Rosellini M, Massafra R, et al. The impact of ECOG performance status on efficacy of immunotherapy and immune-based combinations in cancer patients: the MOUSEION-06 study. Clin Exp Med. 2023;23(8):5039-5049.
doi pubmed - Rizzo A, Brandi G. Neoadjuvant therapy for cholangiocarcinoma: A comprehensive literature review. Cancer Treat Res Commun. 2021;27:100354.
doi pubmed - Rizzo A, Ricci AD, Brandi G. Systemic adjuvant treatment in hepatocellular carcinoma: tempted to do something rather than nothing. Future Oncol. 2020;16(32):2587-2589.
doi pubmed - Rosellini M, Marchetti A, Mollica V, Rizzo A, Santoni M, Massari F. Prognostic and predictive biomarkers for immunotherapy in advanced renal cell carcinoma. Nat Rev Urol. 2023;20(3):133-157.
doi pubmed - Torasawa M, Yoshida T, Yagishita S, Shimoda Y, Shirasawa M, Matsumoto Y, Masuda K, et al. Nivolumab versus pembrolizumab in previously-treated advanced non-small cell lung cancer patients: A propensity-matched real-world analysis. Lung Cancer. 2022;167:49-57.
doi pubmed - Qian S, Wei Z, Yang W, Huang J, Yang Y, Wang J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front Oncol. 2022;12:985363.
doi pubmed pmc - Delbridge AR, Strasser A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015;22(7):1071-1080.
doi pubmed pmc - Shen Q, Li J, Mai J, Zhang Z, Fisher A, Wu X, Li Z, et al. Sensitizing non-small cell lung cancer to BCL-xL-targeted apoptosis. Cell Death Dis. 2018;9(10):986.
doi pubmed pmc - Shibata Y, Hidaka S, Tagawa Y, Nagayasu T. Bcl-2 protein expression correlates with better prognosis in patients with advanced non-small cell lung cancer. Anticancer Res. 2004;24(3b):1925-1928.
pubmed - Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C. Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol. 2003;160(1):53-64.
doi pubmed pmc - Letai A. BH3 domains as BCL-2 inhibitors: prototype cancer therapeutics. Expert Opin Biol Ther. 2003;3(2):293-304.
doi pubmed - Townsend PA, Kozhevnikova MV, Cexus ONF, Zamyatnin AA, Jr., Soond SM. BH3-mimetics: recent developments in cancer therapy. J Exp Clin Cancer Res. 2021;40(1):355.
doi pubmed pmc - Jakubowska MA, Kerkhofs M, Martines C, Efremov DG, Gerasimenko JV, Gerasimenko OV, Petersen OH, et al. ABT-199 (Venetoclax), a BH3-mimetic Bcl-2 inhibitor, does not cause Ca(2+) -signalling dysregulation or toxicity in pancreatic acinar cells. Br J Pharmacol. 2019;176(22):4402-4415.
doi pubmed pmc - Steele TM, Talbott GC, Sam A, Tepper CG, Ghosh PM, Vinall RL. Obatoclax, a BH3 mimetic, enhances cisplatin-induced apoptosis and decreases the clonogenicity of muscle invasive bladder cancer cells via mechanisms that involve the inhibition of pro-survival molecules as well as cell cycle regulators. Int J Mol Sci. 2019;20(6):1285.
doi pubmed pmc - Kim H, Ahn YH, Moon CM, Kang JL, Woo M, Kim M. Lethal effects of mitochondria via microfluidics. Bioeng Transl Med. 2023;8(3):e10461.
doi pubmed pmc - Kim K, Kim KS, Park H, Ye JC, Park Y. Real-time visualization of 3-D dynamic microscopic objects using optical diffraction tomography. Opt Express. 2013;21(26):32269-32278.
doi pubmed - Prabhu D, Mehanna E, Gargesha M, Wen D, Brandt E, van Ditzhuijzen NS, Chamie D, et al. 3D registration of intravascular optical coherence tomography and cryo-image volumes for microscopic-resolution validation. Proc SPIE Int Soc Opt Eng. 2016;9788:9788.
doi pubmed pmc - Giedt RJ, Sprachman MM, Yang KS, Weissleder R. Imaging cellular distribution of Bcl inhibitors using small molecule drug conjugates. Bioconjug Chem. 2014;25(11):2081-2085.
doi pubmed pmc - Park D, Magis AT, Li R, Owonikoko TK, Sica GL, Sun SY, Ramalingam SS, et al. Novel small-molecule inhibitors of Bcl-XL to treat lung cancer. Cancer Res. 2013;73(17):5485-5496.
doi pubmed pmc - Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202-208.
doi pubmed - Kline MP, Rajkumar SV, Timm MM, Kimlinger TK, Haug JL, Lust JA, Greipp PR, et al. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia. 2007;21(7):1549-1560.
doi pubmed - Saha T, Dash C, Jayabalan R, Khiste S, Kulkarni A, Kurmi K, Mondal J, et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat Nanotechnol. 2022;17(1):98-106.
doi pubmed pmc - Trapani JA, Sutton VR. Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr Opin Immunol. 2003;15(5):533-543.
doi pubmed - Alimonti JB, Shi L, Baijal PK, Greenberg AH. Granzyme B induces BID-mediated cytochrome c release and mitochondrial permeability transition. J Biol Chem. 2001;276(10):6974-6982.
doi pubmed - Elena-Real CA, Diaz-Quintana A, Gonzalez-Arzola K, Velazquez-Campoy A, Orzaez M, Lopez-Rivas A, Gil-Caballero S, et al. Cytochrome c speeds up caspase cascade activation by blocking 14-3-3epsilon-dependent Apaf-1 inhibition. Cell Death Dis. 2018;9(3):365.
doi pubmed pmc - Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275(5303):1129-1132.
doi pubmed - Kuznetsov AV, Margreiter R. Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int J Mol Sci. 2009;10(4):1911-1929.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.